Команда підтримки детоксикації Back Clinic. Детоксикація, яка практикується у всьому світі, полягає в тому, щоб відпочити, очистити та підживити тіло зсередини. Видаляючи та усуваючи токсини, живлячи організм корисними поживними речовинами, детоксикація може допомогти захистити вас від хвороб і відновити вашу здатність підтримувати оптимальне здоров’я за допомогою низки методів, включаючи хіропрактику, медитацію тощо. Крім того, детоксикація означає очищення крові.
Це робиться шляхом видалення домішок з крові в печінці, де токсини обробляються для виведення. Організм також виводить токсини через нирки, кишечник, легені, лімфатичну систему та шкіру. Однак, коли ці системи скомпрометовані, а домішки не фільтруються належним чином, здоров’я організму стає скомпрометованим. Тому кожен повинен хоча б раз на рік проводити детоксикацію.
Проте детоксикація для годуючих матерів, дітей та пацієнтів із хронічними дегенеративними захворюваннями, раком або туберкульозом повинна проконсультуватися з лікарем перед початком програми детоксикації. Крім того, проконсультуйтеся зі своїм лікарем, якщо у вас виникли питання щодо детоксикації. Але в сучасному світі в навколишньому середовищі більше токсинів, ніж будь-коли.
Фред Форман – баскетбольний тренер, який залежить від свого загального здоров’я та самопочуття, щоб мати можливість займатися своїми повсякденними обов’язками. В результаті тренер Форман розпочав 6-денна програма детокс від Xymogen, розроблений для того, щоб допомогти оновити та підвищити очищення людського організму та Детоксифікація можливостей.
Фред Форман розповідає про свій досвід 6-денної програми детоксикації, описуючи переваги, які він розробив, а також зусилля, які йому довелося реалізувати, щоб підтримати своє загальне здоров’я та самопочуття за допомогою детоксикації. Фред Форман відчуває велике відчуття задоволення від 6-денної програми детоксикації, і він заохочує інших людей, які також бажають покращити своє самопочуття, детоксикувати своє тіло. Тренер Форман настійно рекомендує 6-денну програму детоксикації як альтернативний вибір лікування загальне самопочуття та самопочуття.
Клініка травм і хіропрактики
Ми благословенні представити вам Клініка для здоров’я та лікування травм Ель-Пасо.
Як клініка хіропрактичної реабілітації та інтегрований медичний центр Ель-Пасо, ми пристрасно зосереджені на лікуванні пацієнтів після неприємних травм і хронічних больових синдромів. Ми зосереджуємось на покращенні ваших здібностей за допомогою програм гнучкості, мобільності та спритності, розроблених для всіх вікових груп та інвалідів.
Ми хочемо, щоб ви жили життям, наповненим більшою енергією, позитивним настроєм, кращим сном, меншим болем, належною вагою тіла та освічені, як підтримувати такий спосіб життя.
Запевняю вас, я прийму лише найкраще для вас
Якщо вам сподобалося це відео, і ми чимось вам допомогли, не соромтеся підписуватися та рекомендуйте нас.
Кетонові тіла виробляються печінкою і використовуються як джерело енергії, коли глюкоза недоступна в організмі людини. Двома основними кетоновими тілами є ацетоацетат (AcAc) і 3-бета-гідроксибутират (3HB), тоді як ацетон є третім і найменш поширеним кетоновим тілом. Кетони завжди присутні в крові, і їх рівень підвищується під час голодування і тривалих фізичних навантажень.�Кетогенез це біохімічний процес, за допомогою якого організми виробляють кетонові тіла шляхом розщеплення жирних кислот і кетогенних амінокислот.
Кетонові тіла в основному утворюються в мітохондрії клітин печінки. Кетогенез відбувається, коли в крові низький рівень глюкози, особливо після того, як інші клітинні запаси вуглеводів, наприклад глікоген, були вичерпані. Цей механізм також може виникнути при недостатній кількості інсуліну. Виробництво кетонових тіл в кінцевому підсумку ініціюється, щоб зробити доступною енергію, яка зберігається в організмі людини у вигляді жирних кислот. Кетогенез відбувається в мітохондріях, де він регулюється незалежно.
абстрактний
Метаболізм кетонових тіл є центральним вузлом фізіологічного гомеостазу. У цьому огляді ми обговорюємо, як кетони виконують дискретні метаболічні функції, які оптимізують роботу органів і організмів у різних залишках поживних речовин і захищають від запалення та травм у багатьох системах органів. Традиційно розглядаються як метаболічні субстрати, які беруть участь лише при обмеженні вуглеводів, нещодавні спостереження підкреслюють важливість кетонових тіл як життєво важливих метаболічних і сигнальних медіаторів, коли вуглеводи є у великій кількості. Доповнюючи репертуар відомих терапевтичних можливостей для захворювань нервової системи, виникла перспективна роль кетонових тіл при раку, а також інтригуючі захисні ролі в серці та печінці, відкриваючи терапевтичні можливості при ожирінні та серцево-судинних захворюваннях. Для узгодження класичної догми з сучасними спостереженнями обговорюються суперечності в метаболізмі кетонів і передачі сигналів.
Вступ
Кетонові тіла є життєво важливим альтернативним джерелом метаболічного палива для всіх сфер життя, еукарії, бактерій та архей (Aneja et al., 2002; Cahill GF Jr, 2006; Krishnakumar et al., 2008). Метаболізм кетонових тіл у людей був використаний для підживлення мозку під час епізодичних періодів нестачі поживних речовин. Кетонові тіла переплетені з ключовими метаболічними шляхами ссавців, такими як ?-окислення (FAO), цикл трикарбонових кислот (TCA), глюконеогенез, de novo ліпогенез (DNL) і біосинтез стеролів. У ссавців кетонові тіла виробляються переважно в печінці з ацетил-КоА, що походить від ФАО, і вони транспортуються до позапечінкових тканин для кінцевого окислення. Ця фізіологія забезпечує альтернативне паливо, яке доповнюється відносно короткими періодами голодування, що збільшує доступність жирних кислот і зменшує доступність вуглеводів (Cahill GF Jr, 2006; McGarry and Foster, 1980; Robinson and Williamson, 1980). Окислення кетонових тіл стає важливим фактором загального енергетичного метаболізму ссавців у позапечінкових тканинах у безлічі фізіологічних станів, включаючи голодування, голодування, період новонародженості, після тренування, вагітність та дотримання дієти з низьким вмістом вуглеводів. У здорових дорослих людей загальні концентрації кетонових тіл у циркуляції зазвичай мають циркадні коливання між приблизно 100-250 мМ, підвищуються до ~1 мМ після тривалих фізичних навантажень або 24-годинного голодування і можуть накопичуватися до 20 мМ при таких патологічних станах, як діабетичний кетоацидоз ( Cahill GF Jr, 2006; Johnson et al., 1969b; Koeslag та ін., 1980; Robinson and Williamson, 1980; Wildenhoff та ін., 1974). Печінка людини виробляє до 300 г кетонових тіл на добу (Balasse and Fery, 1989), які вносять від 5 до 20% загальних витрат енергії при нагодуванні, голодуванні та голодуванні (Balasse et al., 1978; Cox et al. ін., 2016).
Недавні дослідження висвітлюють важливу роль кетонових тіл у метаболізмі клітин ссавців, гомеостазі та передачі сигналів у широкому діапазоні фізіологічних та патологічних станів. Окрім того, що вони служать енергетичним паливом для позапечінкових тканин, таких як мозок, серце або скелетні м’язи, кетонові тіла відіграють ключову роль як сигнальні медіатори, рушії посттрансляційної модифікації білка (PTM) і модулятори запалення та окислювального стресу. У цьому огляді ми надаємо як класичні, так і сучасні погляди на плейотропну роль кетонових тіл та їх метаболізм.
Огляд метаболізму кетонового тіла
Швидкість кетогенезу печінки регулюється організованою серією фізіологічних і біохімічних перетворень жиру. Основні регулятори включають ліполіз жирних кислот з триацилгліцеринів, транспортування до плазматичної мембрани гепатоцитів і через неї, транспортування в мітохондрії за допомогою карнітинпальмітоілтрансферази 1 (CPT1), спіраль β-окислення, активність циклу TCA та проміжні концентрації, потенціал окисно-відновного регулювання та гормональний регуляторний потенціал. з цих процесів, переважно глюкагон та інсулін [огляд в (Arias et al., 1995; Ayte et al., 1993; Ehara et al., 2015; Ferre et al., 1983; Kahn et al., 2005; McGarry and Foster , 1980; Williamson et al., 1969)]. Класично кетогенез розглядають як шлях поширення, при якому ацетил-КоА, отриманий від α-окислення, перевищує активність цитратсинтази та/або доступність оксалоацетату для конденсації з утворенням цитрату. Тривуглецеві проміжні продукти виявляють антикетогенну активність, імовірно, через їхню здатність розширювати пул оксалоацетату для споживання ацетил-КоА, але сама концентрація ацетил-КоА в печінці не визначає швидкість кетогенезу (Foster, 1967; Rawat and Menahan, 1975; Williamson та ін., 1969). Регулювання кетогенезу гормональними, транскрипційними та посттрансляційними подіями разом підтверджує думку про те, що молекулярні механізми, які точно регулюють швидкість кетогенезу, залишаються неповністю зрозумілими (див. Регулювання HMGCS2 і SCOT/OXCT1).
Кетогенез відбувається переважно в мітохондріальному матриксі печінки зі швидкістю, пропорційною до загального окислення жиру. Після транспортування ацильних ланцюгів через мітохондріальні мембрани та ?-окислення мітохондріальна ізоформа 3-гідроксиметилглутарил-КоА-синтази (HMGCS2) каталізує долю, в результаті чого відбувається конденсація ацетоацетил-КоА (AcAc-CoA) і утворення HMG-Ac-CoA. (рис. 1А). HMG-CoA ліаза (HMGCL) розщеплює HMG-CoA з вивільненням ацетил-КоА та ацетоацетату (AcAc), а останній відновлюється до d-?-гідроксибутирату (d-?OHB) за допомогою фосфатидилхолін-залежної мітохондріальної d-?OHB дегідрогенази. BDH1) у близькорівноважній реакції, пов’язаної з NAD+/NADH (Bock and Fleischer, 1975; LEHNINGER et al., 1960). Константа рівноваги BDH1 сприяє виробленню d-?OHB, але співвідношення кетонових тіл AcAc/d-?OHB прямо пропорційне відношенню мітохондріальних NAD+/NADH, і, таким чином, активність оксидоредуктази BDH1 модулює окислювально-відновний потенціал мітохондрій (Krebs et al., 1969; Вільямсон та ін., 1967). AcAc також може спонтанно декарбоксилюватися до ацетону (Pedersen, 1929), джерела солодкого запаху у людей, які страждають на кетоацидоз (тобто загальна кількість кетонових тіл сироватки > ~7 мМ; AcAc pKa 3.6, ?OHB pKa 4.7). Механізми, за допомогою яких кетонові тіла транспортуються через внутрішню мембрану мітохондрій, невідомі, але AcAc/d-?OHB вивільняються з клітин за допомогою монокарбоксилатних транспортерів (у ссавців MCT 1 і 2, також відомі як переносники розчиненої речовини 16A, члени сімейства 1 і 7) і транспортується в кровообігу до позапечінкових тканин для термінального окислення (Cotter et al., 2011; Halestrap and Wilson, 2012; Halestrap, 2012; Hugo et al., 2012). Концентрації циркулюючих кетонових тіл вищі, ніж у позапечінкових тканинах (Harrison and Long, 1940), що вказує на те, що кетонові тіла транспортуються вниз за градієнтом концентрації. Мутації втрати функції в MCT1 пов’язані зі спонтанними нападами кетоацидозу, що свідчить про вирішальну роль в імпорті кетонових тіл.
� За винятком можливого переведення кетонових тіл у неокислювальні долі (див. Неокислювальні метаболічні долі кетонових тіл), гепатоцити не мають здатності метаболізувати кетонові тіла, які вони виробляють. Кетонові тіла, синтезовані печінкою de novo, (i) катаболізуються в мітохондріях позапечінкових тканин до ацетил-КоА, який доступний для циклу TCA для кінцевого окислення (рис. 1A), (ii) направляється на шляхи ліпогенезу або синтезу стеролів ( Рис. 1B), або (iii) виводиться із сечею. Як альтернативне енергетичне паливо, кетонові тіла жадібно окислюються в серці, скелетних м’язах і мозку (Balasse and Fery, 1989; Bentourkia et al., 2009; Owen et al., 1967; Reichard et al., 1974; Sultan, 1988 ). Позапечінковий мітохондріальний BDH1 каталізує першу реакцію окислення ?OHB, перетворюючи його в зворотний AcAc (LEHNINGER et al., 1960; Sandermann et al., 1986). Цитоплазматична d-?OHB-дегідрогеназа (BDH2) з ідентичністю послідовності лише на 20% з BDH1 має високу Km для кетонових тіл, а також відіграє роль у гомеостазі заліза (Davuluri et al., 2016; Guo et al., 2006) . У позапечінковому мітохондріальному матриксі AcAc активується до AcAc-CoA шляхом обміну CoA-частиною з сукциніл-CoA в реакції, що каталізується унікальною CoA трансферазою ссавців, сукциніл-CoA:3-oxoacid-CoA трансферазою (SCOT, CoA transferase; кодується OXCT1) через реакцію, близьку до рівноваги. Вільна енергія, що виділяється при гідролізі AcAc-CoA, більша, ніж сукциніл-CoA, що сприяє утворенню AcAc. Таким чином, окислювальний потік кетонових тіл відбувається внаслідок дії маси: рясний запас AcAc і швидке споживання ацетил-КоА через цитратсинтазу сприяє утворенню AcAc-CoA (+ сукцинат) SCOT. Примітно, що на відміну від глюкози (гексокінази) і жирних кислот (ацил-КоА-синтетаз), активація кетонових тіл (СКОТ) у форму, що окислюється, не вимагає вкладення АТФ. Зворотна реакція тіолази AcAc-CoA [що каталізується будь-якою з чотирьох мітохондріальних тіолаз, що кодуються ACAA2 (кодує фермент, відомий як T1 або CT), ACAT1 (кодує T2), HADHA або HADHB], дає дві молекули ацетил-CoA які входять у цикл TCA (Hersh і Jencks, 1967; Stern et al., 1956; Williamson et al., 1971). Під час кетотичних станів (тобто загальні кетони сироватки > 500 мкМ) кетонові тіла стають значним внеском у витрати енергії і швидко використовуються в тканинах, поки не відбувається поглинання або насичення окислення (Balasse et al., 1978; Balasse and Fery, 1989 Едмонд та ін., 1987). Дуже невелику частку кетонових тіл, отриманих з печінки, можна легко виміряти в сечі, а швидкість утилізації та реабсорбції нирками пропорційна концентрації в крові (Goldstein, 1987; Robinson and Williamson, 1980). Під час високо кетотичних станів (> 1 мМ у плазмі) кетонурія служить напівкількісним звітом про кетоз, хоча більшість клінічних аналізів кетонових тіл у сечі виявляє AcAc, але не ?OHB (Klocker et al., 2013).
Кетогенні субстрати та їх вплив на метаболізм гепатоцитів
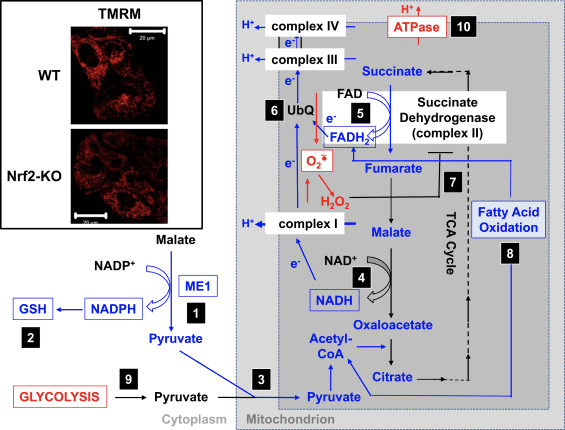
Кетогенні субстрати включають жирні кислоти та амінокислоти (рис. 1В). Катаболізм амінокислот, особливо лейцину, утворює близько 4% кетонових тіл у стані після абсорбції (Thomas et al., 1982). Таким чином, пул субстратів ацетил-КоА для утворення кетонових тіл в основному походить з жирних кислот, оскільки під час станів зниженого постачання вуглеводів піруват потрапляє в печінковий цикл TCA переважно через анаплероз, тобто АТФ-залежне карбоксилування до оксалоацетату (OAA) або до малату. (MAL), а не окисне декарбоксилювання до ацетил-КоА (Jeoung et al., 2012; Magnusson et al., 1991; Merritt et al., 2011). У печінці глюкоза та піруват мізерно сприяють кетогенезу, навіть коли декарбоксилювання пірувату до ацетил-КоА є максимальним (Jeoung et al., 2012).
Ацетил-КоА відіграє кілька ролей, невід'ємних від проміжного метаболізму в печінці, крім генерації АТФ через кінцеве окислення (також див. Інтеграція метаболізму кетонових тіл, посттрансляційної модифікації та фізіології клітин). Ацетил-КоА алостерично активує (i) піруваткарбоксилазу (PC), тим самим активуючи механізм метаболічного контролю, який посилює анаплеротичний надходження метаболітів у цикл TCA (Owen et al., 2002; Scrutton and Utter, 1967) і (ii) дегідрогеназу. кіназа, яка фосфорилює та інгібує піруватдегідрогеназу (PDH) (Cooper et al., 1975), тим самим ще більше посилюючи потік пірувату в цикл TCA через анаплероз. Крім того, цитоплазматичний ацетил-КоА, пул якого збільшується механізмами, які перетворюють мітохондріальний ацетил-КоА в транспортні метаболіти, інгібує окислення жирних кислот: ацетил-КоА-карбоксилаза (ACC) каталізує перетворення ацетил-КоА в малоніл-КоА, ліпогенний субстрат. та алостеричний інгібітор мітохондріального CPT1 [огляд у (Kahn et al., 2005; McGarry and Foster, 1980)]. Таким чином, мітохондріальний пул ацетил-КоА як регулює, так і регулюється переливним шляхом кетогенезу, який керує ключовими аспектами печінкового проміжного метаболізму.
Неокислювальні метаболічні долі кетонових тіл
Переважна доля кетонів, отриманих з печінки, - це SCOT-залежне позапечінкове окислення. Однак AcAc може експортуватися з мітохондрій і використовуватися в анаболічних шляхах шляхом перетворення в AcAc-CoA за допомогою АТФ-залежної реакції, що каталізується цитоплазматичною ацетоацетил-КоА-синтетазою (AACS, рис. 1B). Цей шлях активний під час розвитку мозку та в молочних залозах лактації (Morris, 2005; Robinson and Williamson, 1978; Ohgami et al., 2003). AACS також високо експресується в жировій тканині та активованих остеокластах (Aguilo et al., 2010; Yamasaki et al., 2016). Цитоплазматичний AcAc-CoA може бути або спрямований цитозольним HMGCS1 на біосинтез стеролів, або розщепленим будь-якою з двох цитоплазматичних тіолаз до ацетил-КоА (ACAA1 і ACAT2), карбоксилювати до малоніл-КоА і сприяти синтезу жирних кислот та ін. ін., 1984; Едмонд, 1974; Ендеманн та ін., 1982; Джілен та ін., 1983; Веббер і Едмонд, 1977).
Хоча фізіологічне значення ще не встановлено, кетони можуть служити анаболічним субстратом навіть у печінці. У штучних експериментальних контекстах AcAc може вносити до половини новосинтезованих ліпідів і до 75% нового синтезованого холестерину (Endemann et al., 1982; Geelen et al., 1983; Freed et al., 1988). Оскільки AcAc походить від неповного окислення жирів у печінці, здатність AcAc сприяти ліпогенезу in vivo означатиме безперспективний цикл у печінці, коли кетони, отримані з жиру, можуть бути використані для виробництва ліпідів, поняття, фізіологічне значення якого вимагає експериментального підтвердження, але може служити адаптивні або дезадаптивні ролі (Solinas et al., 2015). AcAc активно забезпечує холестерогенез з низьким вмістом AACS Km-AcAc (~50 мкМ), що сприяє активації AcAc навіть у стані нагодування (Bergstrom et al., 1984). Динамічну роль цитоплазматичного метаболізму кетонів припускають у первинних ембріональних нейронах миші та в адипоцитах, отриманих від 3T3-L1, оскільки нокдаун AACS порушує диференціацію кожного типу клітин (Hasegawa et al., 2012a; Hasegawa et al., ). Нокдаун AACS у мишей in vivo знизив рівень холестерину в сироватці крові (Hasegawa et al., 2012c). SREBP-2012, головний регулятор транскрипції біосинтезу холестерину, і рецептор, активований проліфератором пероксисом (PPAR)-? є активаторами транскрипції AACS і регулюють її транскрипцію під час розвитку нейритів і в печінці (Aguilo et al., 2; Hasegawa et al., 2010c). Взято разом, метаболізм цитоплазматичних кетонових тіл може бути важливим у певних станах або природних історіях захворювань, але недостатній для утилізації кетонових тіл, отриманих з печінки, оскільки масивна гіперкетонемія виникає в умовах селективного порушення первинної окислювальної долі через втрату функціональних мутацій. до SCOT (Berry et al., 2012; Cotter et al., 2001).
Регулювання HMGCS2 і SCOT/OXCT1
Розбіжність мітохондрій від гена, що кодує цитозольний HMGCS, відбулася на ранніх етапах еволюції хребетних через необхідність підтримки печінкового кетогенезу у видів з більш високим співвідношенням маси мозку до тіла (Boukaftane et al., 1994; Cunnane and Crawford, 2003). Природні мутації HMGCS2 з втратою функції у людей викликають напади гіпокетотичної гіпоглікемії (Pitt et al., 2015; Thompson et al., 1997). Надійна експресія HMGCS2 обмежена гепатоцитами та епітелієм товстої кишки, а її експресія та ферментативна активність координуються за допомогою різноманітних механізмів (Mascaro et al., 1995; McGarry and Foster, 1980; Robinson and Williamson, 1980). Хоча повний спектр фізіологічних станів, які впливають на HMGCS2, потребує подальшого з’ясування, його експресія та/або активність регулюються протягом раннього постнатального періоду, старіння, діабету, голодування або вживання кетогенної дієти (Balasse and Fery, 1989; Cahill GF Jr, 2006). ; Girard et al., 1992; Hegardt, 1999; Satapati et al., 2012; Sengupta et al., 2010). У плода метилювання 5� фланкуючої області гена Hmgcs2 обернено корелює з його транскрипцією і частково змінюється після народження (Arias et al., 1995; Ayte et al., 1993; Ehara et al., 2015; Ferre et al. ., 1983). Аналогічно, печінковий Bdh1 демонструє модель експресії розвитку, яка збільшується від народження до відлучення, а також індукується кетогенною дієтою залежно від фактора росту фібробластів (FGF)-21 (Badman et al., 2007; Zhang et al., 1989 ). Кетогенез у ссавців дуже чутливий як до інсуліну, так і до глюкагону, пригнічується і стимулюється відповідно (McGarry and Foster, 1977). Інсулін пригнічує ліполіз жирової тканини, таким чином позбавляючи кетогенез його субстрату, тоді як глюкагон збільшує кетогенний потік через прямий вплив на печінку (Hegardt, 1999). Транскрипцію Hmgcs2 стимулює транскрипційний фактор FOXA2, який інгібується інсулін-фосфатидилінозитол-3-кіназою/Akt і індукується передачі сигналів глюкагон-цАМФ-p300 (Arias et al., 1995; Hegardt, 1999; Quant, 1990). , 1993; Thumelin et al., 2013; von Meyenn et al., 2004; Wolfrum et al., 2003; Wolfrum et al., XNUMX). PPAR? (Rodriguez et al., 1994) разом зі своєю мішенню FGF21 (Badman et al., 2007) також індукує транскрипцію Hmgcs2 в печінці під час голодування або застосування кетогенної дієти (Badman et al., 2007; Inagaki et al., 2007). ). Індукція PPAR? може відбутися до переходу від фізіології плода до новонародженого, тоді як активація FGF21 може сприяти активації в ранньому неонатальному періоді через ?OHB-опосередковане інгібування гістондеацетилази (HDAC)-3 (Rando et al., 2016). mTORC1 (мішень комплексу рапаміцину 1 у ссавців) залежить від інгібування PPAR? транскрипційна активність також є ключовим регулятором експресії гена Hmgcs2 (Sengupta et al., 2010), а PER2 печінки, головний циркадний осцилятор, опосередковано регулює експресію Hmgcs2 (Chavan et al., 2016). Останні спостереження вказують на те, що спричинений позапечінковою пухлиною інтерлейкін-6 погіршує кетогенез через PPAR? придушення (Flint et al., 2016).
Активність ферменту HMGCS2 регулюється кількома PTM. Фосфорилювання серину HMGCS2 посилило його активність in vitro (Grimsrud et al., 2012). Активність HMGCS2 алостерично інгібується сукцинилуванням сукциніл-КоА та залишків лізину (Arias et al., 1995; Hegardt, 1999; Lowe and Tubbs, 1985; Quant et al., 1990; Rardin et al., 2013; 1975; Тумелін та ін., 1993). Сукцинилування залишків лізину HMGCS2, HMGCL та BDH1 в мітохондріях печінки є мішенню для NAD+-залежної деацилази сиртуїну 5 (SIRT5) (Rardin et al., 2013). Активність HMGCS2 також посилюється деацетилюванням лізину SIRT3, і можливо, що перехресні зв’язки між ацетилюванням та сукцинилуванням регулюють активність HMGCS2 (Rardin et al., 2013; Shimazu et al., 2013). Незважаючи на здатність цих PTM регулювати HMGCS2 Km і Vmax, коливання цих PTM ще не були ретельно нанесені на карту і не були підтверджені як механістичні драйвери кетогенезу in vivo.
SCOT експресується у всіх клітинах ссавців, які містять мітохондрії, за винятком клітин гепатоцитів. Важливість активності SCOT і кетолізу була продемонстрована у мишей SCOT-KO, які демонстрували рівномірну летальність через гіперкетонемічну гіпоглікемію протягом 48 годин після народження (Cotter et al., 2011). Тканинно-специфічна втрата SCOT в нейронах або скелетних міоцитах викликає метаболічні порушення під час голодування, але не є смертельною (Cotter et al., 2013b). У людей дефіцит SCOT на ранньому етапі життя проявляється важким кетоацидозом, що викликає летаргію, блювоту та кому (Berry et al., 2001; Fukao et al., 2000; Kassovska-Bratinova et al., 1996; Niezen-Koning et al. , 1997; Saudubray et al., 1987; Snyderman et al., 1998; Tildon and Cornblath, 1972). На клітинному рівні відомо відносно мало про регулятори експресії гена SCOT і білка. Експресія мРНК Oxct1 і білка SCOT і активність зменшуються в кетотичних станах, можливо, через PPAR-залежні механізми (Fenselau and Wallis, 1974; Fenselau and Wallis, 1976; Grinblat et al., 1986; Okuda et al., 1991; Turko et al. ., 2001; Wentz та ін., 2010). При діабетичному кетоацидозі невідповідність між печінковим кетогенезом і позапечінковим окисленням посилюється через порушення активності SCOT. Надмірна експресія інсулінонезалежного транспортера глюкози (GLUT1/SLC2A1) в кардіоміоцитах також пригнічує експресію гена Oxct1 і пригнічує термінальне окислення кетонів у некетотичному стані (Yan et al., 2009). У печінці кількість мРНК Oxct1 пригнічується мікроРНК-122 і метилюванням гістонів H3K27me3, що є очевидним під час переходу від фетального до неонатального періоду (Thorrez et al., 2011). Однак придушення печінкової експресії Oxct1 в постнатальному періоді в першу чергу пояснюється евакуацією Oxct1-експресуючих гемопоетичних попередників з печінки, а не втратою раніше існуючої експресії Oxct1 в термінально диференційованих гепатоцитах. Насправді експресія мРНК Oxct1 і білка SCOT в диференційованих гепатоцитах надзвичайно низька (Orii et al., 2008).
SCOT також регулюється PTM. Фермент гіперацетильований в мозку мишей SIRT3 KO, які також демонструють знижене залежне від AcAc виробництво ацетил-КоА (Dittenhafer-Reed et al., 2015). Неферментативне нітрування залишків тирозину SCOT також послаблює його активність, про що повідомлялося в серцях різних моделей діабетичних мишей (Marcondes et al., 2001; Turko et al., 2001; Wang et al., 2010a). Навпаки, нітрування залишків триптофану збільшує активність SCOT (Br�g�re et al., 2010; Rebrin et al., 2007). Молекулярні механізми специфічного для залишків нітрування або денітрування, призначені для модуляції активності SCOT, можуть існувати і вимагати з’ясування.
Суперечки в позапечінковому кетогенезі
У ссавців первинним кетогенним органом є печінка, і лише гепатоцити та епітеліальні клітини кишечника рясно експресують мітохондріальну ізоформу HMGCS2 (Cotter et al., 2013a; Cotter et al., 2014; McGarry and Foster, 1980; Robinson1980 і Williamson). . Анаеробна бактеріальна ферментація складних полісахаридів дає бутират, який поглинається колоноцитами у ссавців для кінцевого окислення або кетогенезу (Cherbuy et al., 1995), що може відігравати роль у диференціації колоноцитів (Wang et al., 2016). За винятком епітеліальних клітин і гепатоцитів кишечника, HMGCS2 майже відсутній майже у всіх інших клітинах ссавців, але перспектива позапечінкового кетогенезу була підвищена в пухлинних клітинах, астроцитах центральної нервової системи, нирках, підшлунковій залозі. клітини, пігментний епітелій сітківки (RPE) і навіть у скелетних м’язах (Adijanto et al., 2014; Avogaro et al., 1992; El Azzouny et al., 2016; Grabacka et al., 2016; Kang et al., 2015 ; Le Foll et al., 2014; Nonaka et al., 2016; Takagi et al., 2016a; Thevenet et al., 2016; Zhang et al., 2011). Ектопічний HMGCS2 спостерігався в тканинах, яким не вистачає чистої кетогенної здатності (Cook et al., 2016; Wentz et al., 2010), а HMGCS2 проявляє проспективну незалежну від кетогенезу «підготовку» активність, у тому числі всередині клітинного ядра (Chen et al. , 2016; Костюк та ін., 2010; Меертенс та ін., 1998).
Будь-яка позапечінкова тканина, яка окислює кетонові тіла, також може накопичувати кетонові тіла за допомогою незалежних механізмів HMGCS2 (рис. 2A). Однак немає позапечінкової тканини, в якій стабільна концентрація кетонових тіл перевищує концентрацію в циркуляції (Cotter et al., 2011; Cotter et al., 2013b; Harrison and Long, 1940), що підкреслює, що кетонові тіла транспортуються вниз градієнт концентрації через MCT1/2-залежні механізми. Один з механізмів очевидного позапечінкового кетогенезу може насправді відображати відносне порушення окислення кетонів. Додаткові потенційні пояснення належать до сфери утворення кетонових тіл. По-перше, кетогенез de novo може відбуватися через оборотну ферментативну активність тіолази та SCOT (Weidemann and Krebs, 1969). Коли концентрація ацетил-КоА відносно висока, реакції, які зазвичай відповідають за окислення AcAc, відбуваються у зворотному напрямку (GOLDMAN, 1954). Другий механізм виникає, коли проміжні продукти, отримані від ?-окислення, накопичуються через вузьке місце циклу TCA, AcAc-CoA перетворюється на l-?OHB-CoA за допомогою реакції, що каталізується мітохондріальною 3-гідроксиацил-КоА-дегідрогеназою, а також 3-гідроксибутирилом. КоА деацилаза до l-?OHB, яку неможливо відрізнити за допомогою мас-спектрометрії або резонансної спектроскопії від фізіологічного енантіомеру d-?OHB (Reed and Ozand, 1980). l-?OHB можна хроматографічно або ферментативно відрізнити від d-?OHB, і він присутній у позапечінкових тканинах, але не в печінці чи крові (Hsu et al., 2011). Печінковий кетогенез утворює лише d-?OHB, єдиний енантіомер, який є субстратом BDH (Ito et al., 1984; Lincoln et al., 1987; Reed and Ozand, 1980; Scofield et al., 1982; Scofield et al., 1982). Третій HMGCS2-незалежний механізм генерує d-?OHB через катаболізм амінокислот, зокрема лейцину та лізину. Четвертий механізм є очевидним лише тому, що він пов’язаний з артефактом мічення, тому його називають псевдокетогенезом. Це явище пояснюється оборотністю реакцій SCOT і тіолази і може викликати переоцінку обміну кетонових тіл через ізотопне розведення індикатора кетонових тіл у позапечінковій тканині (Des Rosiers et al., 1990; Fink et al., 1988). . Тим не менш, псевдокетогенез може бути незначним у більшості контекстів (Bailey et al., 1990; Keller et al., 1978). Схема (рис. 2A) вказує на корисний підхід, який слід застосувати, розглядаючи підвищену концентрацію кетонів у стабільному стані тканини.
� Нирки нещодавно приділяли увагу як потенційно кетогенний орган. У переважній більшості штатів нирки є чистим споживачем кетонових тіл, отриманих з печінки, виділяють або реабсорбують кетонові тіла з кровотоку, а нирка, як правило, не є чистим генератором або концентратором кетонових тіл (Robinson and Williamson, 1980). Автори класичного дослідження дійшли висновку, що мінімальний нирковий кетогенез, кількісно визначений у штучній експериментальній системі, не має фізіологічного значення (Weidemann and Krebs, 1969). Нещодавно на моделях мишей з цукровим діабетом та дефіцитом аутофагії було зроблено висновок про нирковий кетогенез, але більш імовірно, що багатоорганні зрушення в метаболічному гомеостазі змінюють інтегративний кетоновий метаболізм через введення в різні органи (Takagi et al., 2016a; Takagi et al., 2016b; Zhang et al., 2011). В одній нещодавній публікації було запропоновано нирковий кетогенез як захисний механізм проти ішемічно-реперфузійного ураження нирок (Tran et al., 2016). Повідомлялося про абсолютну рівноважну концентрацію ?OHB з екстрактів ниркової тканини мишей на рівні ~4 мМ. Щоб перевірити, чи це було спроможним, ми кількісно визначили концентрації ?OHB в ниркових екстрактах мишей, які годували та голодували протягом 12 годин. Концентрація ?OHB у сироватці зросла від ~24 мМ до 100 мМ при 2-годинному голодуванні (рис. 24В), у той час як концентрація ?OHB у нирках у рівноважному стані становить приблизно 2 мкМ у стані нагодування і лише 100 мМ у стані 1-годинного голодування (рис. 24C�E), спостереження, які узгоджуються з кількісно визначеними концентраціями понад 2 років тому (Hems and Brosnan, 45). Залишається можливим, що в кетозних станах кетонові тіла, отримані з печінки, можуть бути ренопротекторними, але докази ниркового кетогенезу потребують додаткового обґрунтування. Переконливі докази, що підтверджують справжній позапечінковий кетогенез, були представлені в RPE (Adijanto et al., 1970). Було запропоновано, що ця інтригуюча метаболічна трансформація потенційно дозволить кетонам, отриманим з RPE, надходити до фоторецепторних клітин або клітин глії Меллера, що може сприяти регенерації зовнішнього сегмента фоторецептора.
OHB як посередник сигналізації
Хоча вони енергетично багаті, кетонові тіла виконують провокаційні «неканонічні» сигнальні ролі в клітинному гомеостазі (рис. 3) (Newman and Verdin, 2014; Rojas-Morales et al., 2016). Наприклад, ?OHB інгібує HDAC класу I, що посилює ацетилювання гістонів і, таким чином, індукує експресію генів, які знижують окислювальний стрес (Shimazu et al., 2013). Сам ?OHB є ковалентним модифікатором гістонів на залишки лізину в печінці діабетичних мишей натщесерце або індукованих стрептозотоцином (Xie et al., 2016) (також див. нижче, Інтеграція метаболізму кетонових тіл, посттрансляційної модифікації та фізіології клітин та Кетонові тіла, окислювальний стрес і нейропротекция).
?OHB також є ефектором через рецептори, пов'язані з G-білком. Завдяки незрозумілим молекулярним механізмам він пригнічує активність симпатичної нервової системи та зменшує загальні витрати енергії та частоту серцевих скорочень, пригнічуючи передачу сигналів коротколанцюгових жирних кислот через рецептор, пов’язаний з G-білком 41 (GPR41) (Kimura et al., 2011). Один з найбільш вивчених сигнальних ефектів ?OHB протікає через GPR109A (також відомий як HCAR2), член підродини GPCR гідрокарбонових кислот, що експресується в жировій тканині (білої та коричневої) (Tunaru et al., 2003), і в імунні клітини (Ahmed et al., 2009). ?OHB є єдиним відомим ендогенним лігандом рецептора GPR109A (EC50 ~770 мкМ), активованим d-?OHB, l-?OHB і бутиратом, але не AcAc (Taggart et al., 2005). Високий поріг концентрації для активації GPR109A досягається шляхом дотримання кетогенної дієти, голодування або під час кетоацидозу, що призводить до пригнічення ліполізу жирової тканини. Антиліполітичний ефект GPR109A протікає через інгібування аденілілциклази та зниження цАМФ, інгібуючи чутливу до гормонів тригліцерид-ліпазу (Ahmed et al., 2009; Tunaru et al., 2003). Це створює петлю негативного зворотного зв’язку, в якій кетоз гальмує кетогенез, зменшуючи вивільнення неетерифікованих жирних кислот з адипоцитів (Ahmed et al., 2009; Taggart et al., 2005), ефект, який можна збалансувати симпатичний потяг, який стимулює ліполіз. Ніацин (вітамін B3, нікотинова кислота) є потужним (EC50 ~ 0.1 мкМ) лігандом для GRP109A, ефективно використовуваним протягом десятиліть для лікування дисліпідемій (Benyo et al., 2005; Benyo et al., 2006; Fabbrini et al.;, 2010; Лукасова та ін., 2011; Тунару та ін., 2003). Хоча ніацин посилює зворотний транспорт холестерину в макрофагах і зменшує атеросклеротичні ураження (Lukasova et al., 2011), вплив ?OHB на атеросклеротичні ураження залишається невідомим. Хоча рецептор GPR109A виконує захисну роль, і існують інтригуючі зв’язки між використанням кетогенної дієти при інсульті та нейродегенеративних захворюваннях (Fu et al., 2015; Rahman et al., 2014), захисна роль ?OHB через GPR109A не була продемонстрована in vivo .
Нарешті, ?OHB може впливати на апетит і ситість. Метааналіз досліджень, які вимірювали вплив кетогенних дієт і дієт з дуже низьким вмістом енергії, показав, що учасники, які споживають ці дієти, демонструють вищу ситість порівняно з контрольними дієтами (Gibson et al., 2015). Однак правдоподібним поясненням цього ефекту є додаткові метаболічні або гормональні елементи, які можуть модулювати апетит. Наприклад, миші, які перебували на кетогенній дієті для гризунів, демонстрували підвищені витрати енергії порівняно з мишами, які харчувалися контрольною їжею, незважаючи на подібне споживання калорій, а циркулюючий лептин або гени пептидів, що регулюють харчову поведінку, не змінювалися (Kennedy et al., 2007). Серед запропонованих механізмів, які припускають придушення апетиту за допомогою ?OHB, є як передача сигналів, так і окислення (Laeger et al., 2010). Дослідження специфічної для гепатоцитів делеції гена циркадного ритму (Per2) і імунопреципітації хроматину показали, що PER2 безпосередньо активує ген Cpt1a і опосередковано регулює Hmgcs2, що призводить до порушення кетозу у мишей з нокаутом Per2 (Chavan et al., Chavan et al.). У цих мишей спостерігалося порушення очікування їжі, яке було частково відновлено системним введенням ?OHB. Потрібні подальші дослідження, щоб підтвердити центральну нервову систему як пряму ?OHB-мішень, і чи потрібне окислення кетонів для спостережуваних ефектів, чи задіяний інший сигнальний механізм. Інші дослідники посилалися на можливість локального кетогенезу, отриманого з астроцитів у вентромедіальному гіпоталамусі, як на регулятор споживання їжі, але ці попередні спостереження також будуть корисними від генетичних оцінок та оцінок на основі потоків (Le Foll et al., 2016). Зв'язок між кетозом і нестачею поживних речовин залишається цікавим, оскільки голод і насичення є важливими елементами невдалих спроб схуднути.
Інтеграція метаболізму кетонового тіла, посттрансляційної модифікації та фізіології клітини
Кетонові тіла сприяють утворенню розділених пулів ацетил-КоА, ключового проміжного продукту, який відіграє важливу роль у клітинному метаболізмі (Pietrocola et al., 2015). Одна з функцій ацетил-КоА полягає в тому, щоб служити субстратом для ацетилювання, ковалентної модифікації гістонів, що каталізується ферментами (Choudhary et al., 2014; Dutta et al., 2016; Fan et al., 2015; Menzies et al., 2016 ). Велика кількість динамічно ацетильованих мітохондріальних білків, багато з яких можуть виникати за допомогою неферментативних механізмів, також з'явилася в результаті обчислювальних протеомічних досліджень (Dittenhafer-Reed et al., 2015; Hebert et al., 2013; Rardin et al., 2013 ; Shimazu et al., 2010). Лізиндеацетилази використовують кофактор цинку (наприклад, нуклеоцитозольні HDAC) або NAD+ як сусубстрат (сиртуїни, SIRT) (Choudhary et al., 2014; Menzies et al., 2016). Ацетилпротеом служить одночасно датчиком і ефектором загального клітинного пулу ацетил-КоА, оскільки фізіологічні та генетичні маніпуляції призводять до неферментативних глобальних варіацій ацетилювання (Weinert et al., 2014). Оскільки внутрішньоклітинні метаболіти служать модуляторами ацетилювання залишків лізину, важливо враховувати роль кетонових тіл, чисельність яких дуже динамічна.
?OHB є епігенетичним модифікатором принаймні за двома механізмами. Підвищені рівні ?OHB, викликані голодуванням, обмеженням калорій, прямим прийомом або тривалими фізичними вправами провокують інгібування HDAC або активацію гістонацетилтрансферази (Marosi et al., 2016; Sleiman et al., 2016) або окислювальний стрес (Shimazu et al., 2013) 3. . Інгібування ?OHB HDAC2016 може регулювати метаболічну фізіологію новонароджених (Rando et al., 2016). Незалежно, сам ?OHB безпосередньо модифікує залишки лізину гістонів (Xie et al., XNUMX). Тривале голодування або діабетичний кетоацидоз, викликаний стептозотоцином, посилює ?-гідроксибутирилювання гістонів. Хоча кількість сайтів α-гідроксибутирилування та ацетилювання лізину була порівнянною, спостерігалося стехіометричне β-гідроксибутирилування гістонів, ніж ацетилювання. На окремі гени вплинуло α-гідроксибутирилування гістону лізину проти ацетилювання або метилювання, що свідчить про різні клітинні функції. Невідомо, чи є ?-гідроксибутирилювання спонтанним чи ферментативним, але розширює діапазон механізмів за рахунок кетонових тіл, які динамічно впливають на транскрипцію.
Основні події перепрограмування клітин під час обмеження калорій і нестачі поживних речовин можуть бути опосередковані в SIRT3- та SIRT5-залежному мітохондріальному деацетилюванні та десукцинилуванні, відповідно, регулюючи кетогенні та кетолітичні білки на посттрансляційному рівні в печінці та позапечінкових тканинах (ReDittenhafer al. 2015; Hebert et al., 2013; Rardin et al., 2013; Shimazu et al., 2010). Незважаючи на те, що стехіометричне порівняння зайнятих ділянок не обов’язково безпосередньо пов’язане зі зрушеннями метаболічного потоку, ацетилювання мітохондрій є динамічним і може обумовлюватися концентрацією ацетил-КоА або мітохондріальним pH, а не ферментативними ацетилтрансферазами (Wagner and Payne, 2013). Те, що SIRT3 і SIRT5 модулюють активність ферментів, що метаболізують кетонові тіла, викликає питання про взаємну роль кетонів у формуванні ацетилпротеома, сукцинілпротеома та інших динамічних клітинних мішеней. Дійсно, оскільки варіації кетогенезу відображають концентрації NAD+, виробництво та кількість кетону можуть регулювати активність сиртуїну, впливаючи таким чином на загальні пули ацетил-КоА/сукциніл-КоА, ацилпротеом і, таким чином, фізіологію мітохондрій і клітин. ?-гідроксибутирилювання залишків ферменту лізину може додати ще один шар до клітинного перепрограмування. У позапечінкових тканинах окислення кетонових тіл може стимулювати аналогічні зміни гомеостазу клітин. У той час як компартментація пулів ацетил-КоА дуже регулюється і координує широкий спектр клітинних змін, здатність кетонових тіл безпосередньо формувати концентрацію ацетил-КоА в мітохондріях і цитоплазмі потребує з’ясування (Chen et al., 2012; Corbet et al., 2016; Поуговкіна та ін., 2014; Швер та ін., 2009; Wellen and Thompson, 2012). Оскільки концентрації ацетил-КоА жорстко регулюються, а ацетил-КоА є непроникним для мембрани, важливо враховувати механізми, що координують гомеостаз ацетил-КоА, включаючи швидкість виробництва та кінцевого окислення в циклі TCA, перетворення в кетонові тіла, мітохондріальні відтік через карнітин-ацетилтрансферазу (CrAT) або експорт ацетил-КоА в цитозоль після перетворення в цитрат і вивільнення за допомогою АТФ-цитрат-ліази (ACLY). Ключова роль цих останніх механізмів у клітинному ацетилпротеомі та гомеостазі вимагає відповідного розуміння ролі кетогенезу та окислення кетонів (Das et al., 2015; McDonnell et al., 2016; Moussaieff et al., 2015; Overmyer et al., 2015; Seiler et al., 2014; Seiler et al., 2015; Wellen et al., 2009; Wellen and Thompson, 2012). Для визначення цілей та результатів знадобляться конвергентні технології в метаболоміці та ацилпротеоміці в умовах генетично маніпульованих моделей.
Проти- та прозапальні реакції на кетонові тіла
Кетоз і кетонові тіла модулюють запалення і функцію імунних клітин, але були запропоновані різноманітні і навіть невідповідні механізми. Тривала нестача поживних речовин зменшує запалення (Youm et al., 2015), але хронічний кетоз діабету 1 типу є прозапальним станом (Jain et al., 2002; Kanikarla-Marie and Jain, 2015; Kurepa et al., 2012). ). Сигнальні ролі для ?OHB у запаленні з’являються на основі механізмів, оскільки багато клітин імунної системи, включаючи макрофаги або моноцити, рясно експресують GPR109A. Хоча ?OHB має переважно протизапальну відповідь (Fu et al., 2014; Gambhir et al., 2012; Rahman et al., 2014; Youm et al., 2015), високі концентрації кетонових тіл, зокрема AcAc, можуть викликати прозапальну відповідь (Jain et al., 2002; Kanikarla-Marie and Jain, 2015; Kurepa et al., 2012).
Було розглянуто протизапальну роль лігандів GPR109A при атеросклерозі, ожирінні, запальних захворюваннях кишечника, неврологічних захворюваннях та раку (Graff et al., 2016). Експресія GPR109A посилюється в клітинах RPE моделей діабету, пацієнтів з діабетом (Gambhir et al., 2012) і в мікроглії під час нейродегенерації (Fu et al., 2014). Протизапальні ефекти ?OHB посилюються через надмірну експресію GPR109A в клітинах RPE і скасовуються фармакологічним інгібуванням або генетичним нокаутом GPR109A (Gambhir et al., 2012). ?OHB і екзогенна нікотинова кислота (Taggart et al., 2005), обидва надають протизапальну дію на TNF? або запалення, спричинене LPS шляхом зниження рівня прозапальних білків (iNOS, COX-2) або секретованих цитокінів (TNF?, IL-1?, IL-6, CCL2/MCP-1), частково через інгібування NF -?B транслокація (Fu et al., 2014; Gambhir et al., 2012). ?OHB зменшує ER-стрес і інфламасому NLRP3, активуючи відповідь на антиоксидантний стрес (Bae et al., 2016; Youm et al., 2015). Однак при нейродегенеративному запаленні GPR109A-залежний ?OHB-опосередкований захист не включає медіатори запалення, такі як передача сигналів MAPK (наприклад, ERK, JNK, p38) (Fu et al., 2014), але може знадобитися ЦОГ-1-залежний PGD2 виробництво (Rahman et al., 2014). Цікаво, що макрофаг GPR109A необхідний для прояву нейропротекторного ефекту в моделі ішемічного інсульту (Rahman et al., 2014), але здатність ?OHB пригнічувати запалення NLRP3 у макрофагах, отриманих з кісткового мозку, не залежить від GPR109A та ін. ., 2015). Хоча більшість досліджень пов’язують ?OHB з протизапальною дією, ?OHB може бути прозапальним і підвищувати маркери перекисного окислення ліпідів у гепатоцитах теляти (Shi et al., 2014). Таким чином, протизапальні ефекти ?OHB можуть залежати від типу клітин, концентрації ?OHB, тривалості впливу та наявності чи відсутності співмодуляторів.
На відміну від ?OHB, AcAc може активувати прозапальну передачу сигналів. Підвищений AcAc, особливо при високій концентрації глюкози, посилює пошкодження ендотеліальних клітин через механізм, що залежить від НАДФН-оксидази/оксидативного стресу (Kanikarla-Marie and Jain, 2015). Високі концентрації AcAc у пуповині матерів з цукровим діабетом корелювали з вищою швидкістю окислення білка та концентрацією MCP-1 (Kurepa et al., 2012). Високий AcAc у пацієнтів з цукровим діабетом корелював з TNF? експресія (Jain et al., 2002) і AcAc, але не ?OHB, індукували експресію TNF?, MCP-1, накопичення АФК та знижували рівень цАМФ в клітинах моноцитів людини U937 (Jain et al., 2002; Kurepa et al. ., 2012).
Сигнальні явища, залежні від кетонових тіл, часто викликаються лише при високій концентрації кетонових тіл (> 5 мМ), а у випадку багатьох досліджень, які пов’язують кетони з про- або протизапальними ефектами, через незрозумілі механізми. Крім того, через суперечливий вплив ?OHB проти AcAc на запалення та здатність співвідношення AcAc/?OHB впливати на мітохондріальний окислювально-відновний потенціал, найкращі експерименти, що оцінюють роль кетонових тіл на клітинні фенотипи, порівнюють ефекти AcAc і ? OHB у різних співвідношеннях і в різних кумулятивних концентраціях [наприклад, (Saito et al., 2016)]. Нарешті, AcAc можна придбати в комерційних цілях лише у вигляді літієвої солі або у вигляді етилового ефіру, який вимагає гідролізу основи перед використанням. Катіон літію незалежно індукує каскади передачі сигналу (Manji et al., 1995), а аніон AcAc є лабільним. Нарешті, дослідження з використанням рацемічного d/l-?OHB можуть бути заплутаними, оскільки лише стереоізомер d-?OHB може бути окислений до AcAc, але d-?OHB і l-?OHB можуть кожен сигналізувати через GPR109A, інгібувати запалення NLRP3, і служать ліпогенними субстратами.
Кетонові тіла, окислювальний стрес і нейропротекція
Окислювальний стрес, як правило, визначається як стан, в якому АФК представлені в надлишку через надмірну продукцію та/або порушення елімінації. Ролі кетонових тіл, що пом’якшують антиоксидантний та окислювальний стрес, широко описані як in vitro, так і in vivo, особливо в контексті нейропротекції. Оскільки більшість нейронів не ефективно виробляють високоенергетичні фосфати з жирних кислот, але окислюють кетонові тіла, коли вуглеводів не вистачає, нейропротекторний ефект кетонових тіл особливо важливий (Cahill GF Jr, 2006; Edmond et al., 1987; Yang та ін., 1987). У моделях окисного стресу індукція BDH1 і пригнічення SCOT припускають, що метаболізм кетонових тіл можна перепрограмувати для підтримки різноманітних клітинних сигналів, окислювально-відновного потенціалу або метаболічних потреб (Nagao et al., 2016; Tieu et al., 2003).
Кетонові тіла знижують ступінь пошкодження клітин, пошкодження, смерть та зниження апоптозу в нейронах і кардіоміоцитах (Haces et al., 2008; Maalouf et al., 2007; Nagao et al., 2016; Tieu et al., 2003). Викликані механізми різноманітні і не завжди лінійно пов’язані з концентрацією. Низькі мілімолярні концентрації (d або l)-?OHB поглинають АФК (гідроксильний аніон), тоді як AcAc поглинає численні види АФК, але лише в концентраціях, які перевищують фізіологічний діапазон (IC50 20 мМ) (Haces et al., 67) . І навпаки, сприятливий вплив на окислювально-відновний потенціал ланцюга транспорту електронів є механізмом, який зазвичай пов’язаний з d-?OHB. Хоча всі три кетонових тіла (d/l-?OHB і AcAc) зменшували загибель нейронних клітин і накопичення АФК, викликане хімічним пригніченням гліколізу, лише d-?OHB і AcAc запобігали зниженню АТФ нейронів. І навпаки, у гіпоглікемічній моделі in vivo (d або l)-?OHB, але не AcAc запобігали перекисному окисненню ліпідів гіпокампу (Haces et al., 2008; Maalouf et al., 2008; Marosi et al., 2007; Murphy, 2016 ; Tieu та ін., 2009). Дослідження in vivo на мишах, яких годували кетогенною дієтою (2003% ккал жиру і 87% білка), показали нейроанатомічні варіації антиоксидантної здатності (Ziegler et al., 13), де найбільш глибокі зміни спостерігалися в гіпокампі з підвищенням глутатіонпероксидази та загального антиоксидантні можливості.
Кетогенна дієта, кетонові ефіри (також див. Терапевтичне використання кетогенної дієти та екзогенних кетонових тіл) або введення ?OHB надають нейропротекцію на моделях ішемічного інсульту (Rahman et al., 2014); хвороба Паркінсона (Tieu et al., 2003); напад кисневої токсичності центральної нервової системи (D'Agostino et al., 2013); епілептичні спазми (Yum et al., 2015); мітохондріальна енцефаломіопатія, синдром лактоацидозу та інсультоподібних (MELAS) епізодів (Frey et al., 2016) і хвороби Альцгеймера (Cunnane and Crawford, 2003; Yin et al., 2016). І навпаки, нещодавня доповідь продемонструвала гістопатологічні докази нейродегенеративного прогресування за допомогою кетогенної дієти на моделі аномальної репарації мітохондріальної ДНК трансгенної миші, незважаючи на збільшення мітохондріального біогенезу та антиоксидантних сигнатур (Lauritzen et al., 2016). Інші суперечливі повідомлення свідчать про те, що вплив високих концентрацій кетонових тіл викликає окислювальний стрес. Високі дози ?OHB або AcAc викликали секрецію оксиду азоту, перекисне окислення ліпідів, знижену експресію SOD, глутатіонпероксидази і каталази в гепатоцитах теляти, тоді як у гепатоцитах щурів індукція шляху MAPK приписується AcAc, але не ?OHB (Abdelmegeed et al. ; Ши та ін., 2004; Ши та ін., 2014).
У сукупності більшість звітів пов’язують ?OHB з ослабленням окислювального стресу, оскільки його введення пригнічує вироблення АФК/супероксиду, запобігає перекисному окисленню ліпідів і окислення білка, підвищує рівень антиоксидантного білка та покращує мітохондріальне дихання та виробництво АТФ (Abdelmegeed et al., 2004); Haces et al., 2008; Jain et al., 1998; Jain et al., 2002; Kanikarla-Marie and Jain, 2015; Maalouf та ін., 2007; Maalouf і Rho, 2008; Marosi та ін., 2016; Tie та ін., 2003; Yin та ін., 2016; Ziegler та ін., 2003). Хоча AcAc був більш прямим, ніж ?OHB, пов’язаний з індукцією окисного стресу, ці ефекти не завжди легко відокремити від потенційних прозапальних реакцій (Jain et al., 2002; Kanikarla-Marie and Jain, 2015; Kanikarla-Marie та Джейн, 2016). Крім того, важливо враховувати, що очевидна антиоксидантна користь, яку надають плейотропні кетогенні дієти, не може бути перетворена самими кетоновими тілами, а нейропротекція, яка надається кетоновими тілами, не може бути повністю пов’язана з окислювальним стресом. Наприклад, під час депривації глюкози, в моделі депривації глюкози в нейронах кори, ?OHB стимулював аутофагічний потік і запобігав накопиченню аутофагосом, що було пов’язано зі зниженням загибелі нейронів (Camberos-Luna et al., 2016). d-?OHB також індукує канонічні антиоксидантні білки FOXO3a, SOD, MnSOD і каталазу, проспективно через інгібування HDAC (Nagao et al., 2016; Shimazu et al., 2013).
Неалкогольна жирова хвороба печінки (НАЖХП) і метаболізм кетонового тіла
НАЖХП, асоційована з ожирінням, і неалкогольний стеатогепатит (НАСГ) є найпоширенішими причинами захворювань печінки в західних країнах (Rinella and Sanyal, 2016), а печінкова недостатність, спричинена НАСГ, є однією з найпоширеніших причин для трансплантації печінки. У той час як надлишок триацилгліцеролів у гепатоцитах >5% маси печінки (NAFL) сам по собі не викликає дегенеративної функції печінки, прогресування НАЖХП у людей корелює із системною резистентністю до інсуліну та підвищеним ризиком діабету 2 типу та може сприяти патогенезу захворювання. серцево-судинні захворювання та хронічні захворювання нирок (Fabbrini et al., 2009; Targher et al., 2010; Targher and Byrne, 2013). Патогенні механізми НАЖХП і НАСГ вивчені не повністю, але включають порушення метаболізму гепатоцитів, аутофагію гепатоцитів і стрес ендоплазматичного ретикулума, функцію імунних клітин печінки, запалення жирової тканини та системні медіатори запалення (Fabbrini et al., 2009, Masaniu2013, 2010; ; Targher et al., 2010; Yang et al., 2012). Порушення метаболізму вуглеводів, ліпідів та амінокислот виникають у людей та модельних організмів і сприяють ожирінню, діабету та НАЖХП [огляд у (Farese et al., 2011; Lin and Accili, 2012; Newgard, 2012; Samuel and Шульман, 2013; Sun and Lazar, 2010)]. У той час як аномалії гепатоцитів у метаболізмі ліпідів цитоплазми зазвичай спостерігаються при НАЖХП (Fabbrini et al., 2016b), роль мітохондріального метаболізму, який регулює окислювальне видалення жирів, менш ясна в патогенезі НАЖХП. Аномалії мітохондріального метаболізму виникають і сприяють патогенезу НАЖХП/НАСГ (Hyotylainen et al., 2011; Serviddio et al., 2008; Serviddio et al., 2008; Wei et al., 1974). Існує загальне (Felig et al., 2010; Iozzo et al., 2015; Koliaki et al., 2015; Satapati et al., 2012; Satapati et al., 2011; Sunny et al., 2013), але не однорідне ( Koliaki and Roden, 2016; Perry et al., 2010; Rector et al., 2015) консенсус про те, що до розвитку добросовісного НАСГ окислення мітохондрій печінки і, зокрема окислення жиру, посилюється при ожирінні, системній інсулінорезистентності. і НАЖХП. Цілком імовірно, що в міру прогресування НАЖХП з’являється гетерогенність окислювальної здатності, навіть серед окремих мітохондрій, і в кінцевому підсумку окислювальна функція порушується (Koliaki et al., 2010; Rector et al., 2008; Satapati et al., 2012; Satapati et al. ., XNUMX).
Кетогенез часто використовується як проксі для окислення печінкового жиру. Порушення кетогенезу виникають у міру прогресування НАЖХП на тваринних моделях і, ймовірно, у людей. Через неповно визначені механізми гіперінсулінемія пригнічує кетогенез, що, можливо, сприяє гіпокетонемії в порівнянні з контрольною групою з худим організмом (Bergman et al., 2007; Bickerton et al., 2008; Satapati et al., 2012; Soeters et al., Sunny et al., Sunny , 2009; Vice та ін., 2011). Тим не менш, здатність концентрації циркулюючих кетонових тіл передбачати НАЖХП є суперечливою (M�nnist� et al., 2005; Sanyal et al., 2015). Надійні кількісні магнітно-резонансні спектроскопічні методи на тваринних моделях виявили збільшення швидкості обміну кетонів при помірній інсулінорезистентності, але зниження показників було очевидним при більш вираженій інсулінорезистентності (Satapati et al., 2001; Sunny et al., 2012). У людей з ожирінням із жировою печінкою швидкість кетогенезу є нормальною (Bickerton et al., 2010; Sunny et al., 2008), і, отже, швидкість кетогенезу зменшується порівняно зі збільшенням навантаження жирних кислот у гепатоцитах. Отже, ацетил-КоА, отриманий від ?-окислення, може бути спрямований на кінцеве окислення в циклі ТСА, посилюючи термінальне окислення, глюконеогенез, керований фосфоенолпіруватом через анаплероз/катаплероз, і окислювальний стрес. Ацетил-КоА також, можливо, експортується з мітохондрій у вигляді цитрату, субстрату-попередника для ліпогенезу (рис. 2011) (Satapati et al., 4; Satapati et al., 2015; Solinas et al., 2012). Хоча кетогенез стає менш чутливим до інсуліну або голодування при тривалому ожирінні (Satapati et al., 2015), основні механізми та наслідки цього залишаються неповністю зрозумілими. Останні дані вказують на те, що mTORC2012 пригнічує кетогенез таким чином, що може бути нижче за передачу сигналів інсуліну (Kucejova et al., 1), що узгоджується з спостереженнями про те, що mTORC2016 пригнічує PPAR?-опосередковану індукцію Hmgcs1 (Sengupta et al.), 2 також див. Регламент HMGCS2010 і SCOT/OXCT2).
Попередні спостереження нашої групи свідчать про несприятливі наслідки кетогенної недостатності для печінки (Cotter et al., 2014). Щоб перевірити гіпотезу про те, що порушення кетогенезу, навіть у багатих вуглеводами і, таким чином, «некетогенних» станах сприяє аномальному метаболізму глюкози та провокує стеатогепатит, ми створили мишачу модель вираженої кетогенної недостатності шляхом введення антисмислового олігонуклеї-мішені (ASO). Hmgcs2. Втрата HMGCS2 у дорослих мишей, яких годували стандартними їжею з низьким вмістом жиру, спричинила легку гіперглікемію та помітно збільшила продукцію сотень печінкових метаболітів, набір яких вказував на активацію ліпогенезу. Харчування мишей з високим вмістом жиру з недостатнім кетогенезом призводило до обширного пошкодження та запалення гепатоцитів. Ці висновки підтверджують головну гіпотезу про те, що (i) кетогенез не є пасивним шляхом переповнення, а скоріше динамічним вузлом у печінковому та інтегрованому фізіологічному гомеостазі, і (ii) заслуговує на дослідження розумне кетогенне збільшення для пом’якшення НАЖХП/НАСГ та порушення метаболізму глюкози в печінці. .
Як порушення кетогенезу може сприяти ураженню печінки та зміненому гомеостазу глюкози? Перше міркування полягає в тому, чи виною є дефіцит кетогенного потоку, чи самі кетони. Нещодавня доповідь свідчить про те, що кетонові тіла можуть пом’якшити пошкодження печінки, викликане окислювальним стресом, у відповідь на n-3 поліненасичені жирні кислоти (Pawlak et al., 2015). Нагадаємо, що через відсутність експресії SCOT в гепатоцитах кетонові тіла не окислюються, але вони можуть сприяти ліпогенезу та виконувати різноманітні сигнальні ролі незалежно від їх окислення (також див. Неокислювальні метаболічні долі кетонових тіл і ?OHB як сигнальний посередник). Також можливо, що кетонові тіла, отримані з гепатоцитів, можуть служити сигналом та/або метаболітом для сусідніх типів клітин у ацинусах печінки, включаючи зірчасті клітини та макрофаги клітин Купфера. Хоча обмежена доступна література свідчить про те, що макрофаги не здатні окислювати кетонові тіла, це було виміряно лише за допомогою класичних методів і лише в перитонеальних макрофагах (Newsholme et al., 1986; Newsholme et al., 1987), що вказує на те, що повторна оцінка доцільна з огляду на значну експресію SCOT в макрофагах, отриманих з кісткового мозку (Youm et al., 2015).
Кетогенний потік гепатоцитів також може бути цитопротекторним. Хоча цілющі механізми можуть не залежати від кетогенезу як такого, кетогенні дієти з низьким вмістом вуглеводів пов’язують із поліпшенням НАЖХП (Browning et al., 2011; Foster et al., 2010; Kani et al., 2014; Schugar and Crawford, 2012). . Наші спостереження показують, що кетогенез гепатоцитів може регулювати потік циклу TCA, анаплеротичний потік, глюконеогенез, отриманий з фосфоенолпірувату (Cotter et al., 2014), і навіть обмін глікогену. Кетогенне порушення спрямовує ацетил-КоА на збільшення потоку ТЦА, що в печінці пов’язують із збільшенням ушкодження, опосередкованого АФК (Satapati et al., 2015; Satapati et al., 2012); примушує переведення вуглецю в де ново синтезовані види ліпідів, які можуть виявитися цитотоксичними; і запобігає повторне окислення NADH до NAD+ (Cotter et al., 2014) (рис. 4). У сукупності необхідні майбутні експерименти, щоб розглянути механізми, завдяки яким відносна кетогенна недостатність може стати дезадаптивною, сприяти гіперглікемії, спровокувати стеатогепатит і чи діють ці механізми при НАЖХП/НАСГ людини. Епідеміологічні дані свідчать про порушення кетогенезу під час прогресування стеатогепатиту (Embade et al., 2016; Marinou et al., 2011; M�nnist� et al., 2015; Pramfalk et al., 2015; Safaei et al.), 2016 лікування, яке посилює кетогенез печінки, може виявитися корисним (Degirolamo et al., 2016; Honda et al., 2016).
Кетонові тіла та серцева недостатність (СН)
З швидкістю обміну речовин, що перевищує 400 ккал/кг/добу, і оборотом 6 кг АТФ/добу, серце є органом з найбільшою витратою енергії та окислювальною потребою (Ashrafian et al., 35; Wang et al., 2007б). Переважна більшість обороту енергії міокарда знаходиться в мітохондріях, і 2010% цього запасу надходить від ФАО. Серце є всеїдним і гнучким за нормальних умов, але серце, що патологічно ремоделює (наприклад, внаслідок гіпертонії або інфаркту міокарда), і діабетичне серце стають метаболічно негнучкими (Balasse and Fery, 70; BING, 1989; Fukao et al., 1954). ; Lopaschuk et al., 2004; Taegtmeyer et al., 2010; Taegtmeyer et al., 1980; Young et al., 2002). Справді, генетично запрограмовані аномалії метаболізму серцевого палива на моделях мишей провокують кардіоміопатію (Carley et al., 2002; Neubauer, 2014). У фізіологічних умовах нормальне серце окислює кетонові тіла пропорційно їх доставці за рахунок окислення жирних кислот і глюкози, а міокард є найбільшим споживачем кетонового тіла на одиницю маси (BING, 2007; Crawford et al., 1954; GARLAND et al. ., 2009; Hasselbaink et al., 1962; Jeffrey et al., 2003; Pelletier et al., 1995; Tardif et al., 2007; Yan et al., 2001). У порівнянні з окисленням жирних кислот, кетонові тіла більш енергетично ефективні, дають більше енергії для синтезу АТФ на молекулу кисню, що вкладається (відношення P/O) (Kashiwaya et al., 2009; Sato et al., 2010; Veech, 1995) . Окислення кетонових тіл також дає потенційно вищу енергію, ніж ФАО, підтримуючи убіхінон окисленим, що збільшує окисно-відновний діапазон у ланцюзі транспортування електронів і робить більше енергії доступним для синтезу АТФ (Sato et al., 2004; Veech, 1995). Окислення кетонових тіл також може знизити продукцію АФК і, таким чином, окислювальний стрес (Veech, 2004).
Попередні інтервенційні та обсерваційні дослідження вказують на потенційну оздоровчу роль кетонових тіл у серці. У контексті експериментального ураження ішемії/реперфузії кетонові тіла надавали потенційний кардіопротекторний ефект (Al-Zaid et al., 2007; Wang et al., 2008), можливо, через збільшення кількості мітохондрій у серці або посилення ключового окисного фосфорилювання. медіатори (Snorek et al., 2012; Zou et al., 2002). Недавні дослідження показують, що використання кетонових тіл збільшується в серцях мишей (Aubert et al., 2016) і людей (Bedi et al., 2016), що підтверджує попередні спостереження за людьми (BING, 1954; Fukao et al., 2000); Janardhan et al., 2011; Longo et al., 2004; Rudolph and Schinz, 1973; Tildon and Cornblath, 1972). Концентрації циркулюючих кетонових тіл збільшуються у пацієнтів із серцевою недостатністю, прямо пропорційно тиску наповнення, спостереження, механізм і значення яких залишаються невідомими (Kupari et al., 1995; Lommi et al., 1996; Lommi et al., 1997; Neely et al. ., 1972), але миші з селективним дефіцитом SCOT в кардіоміоцитах демонструють прискорене патологічне ремоделювання шлуночків і сигнатури АФК у відповідь на хірургічне спричинене перевантаженням тиском (Schugar et al., 2014).
Останні інтригуючі спостереження в лікуванні діабету виявили потенційний зв’язок між метаболізмом кетонів у міокарді та патологічним ремоделюванням шлуночків (рис. 5). Інгібування ниркового проксимального канальця котранспортера натрію/глюкози 2 (SGLT2i) збільшує концентрацію циркулюючих кетонових тіл у людей (Ferrannini et al., 2016a; Inagaki et al., 2015) і мишей (Suzuki et al., 2014). кетогенез печінки (Ferrannini et al., 2014; Ferrannini et al., 2016a; Katz and Leiter, 2015; Mudaliar et al., 2015). Вражаюче, що принаймні один із цих препаратів зменшив кількість госпіталізації при СН (наприклад, як виявлено в дослідженні EMPA-REG OUTOME) та покращив серцево-судинну смертність (Fitchett et al., 2016; Sonesson et al., 2016; Wu et al., 2016a Зінман та ін., 2015). Незважаючи на те, що рушійні механізми, що стоять за сприятливими результатами HF для пов’язаного SGLT2i, залишаються предметом активних обговорень, перевага виживання, ймовірно, багатофакторна, в перспективі включає кетоз, але також благотворний вплив на вагу, кров’яний тиск, рівень глюкози та сечової кислоти, жорсткість артерій, симпатичну нервову систему, осмотичні діурез/зменшення об’єму плазми та підвищення гематокриту (Raz and Cahn, 2016; Vallon and Thomson, 2016). Взято разом, думка про те, що терапевтично наростаюча кетонемія у пацієнтів із СН або у пацієнтів із високим ризиком розвитку СН, залишається спірною, але активно досліджується в доклінічних та клінічних дослідженнях (Ferrannini et al., 2016b; Kolwicz et al., 2016; Лопащук та Верма, 2016; Мудаляр та ін., 2016; Тегтмайер, 2016).
Кетонові тіла в біології раку
Зв'язки між кетоновими тілами і раком швидко з'являються, але дослідження як на тваринах, так і на людях дали різні висновки. Оскільки кетоновий метаболізм динамічний і реагує на стан поживних речовин, є привабливим шукати біологічні зв’язки з раком через потенціал для точних методів харчування. Ракові клітини зазнають метаболічного перепрограмування для підтримки швидкої проліферації та росту клітин (DeNicola and Cantley, 2015; Pavlova and Thompson, 2016). Класичний ефект Варбурга в метаболізмі ракових клітин виникає через домінуючу роль гліколізу та молочнокислого бродіння для передачі енергії та компенсації меншої залежності від окисного фосфорилювання та обмеженого мітохондріального дихання (De Feyter et al., 2016; Grabacka et al., 2016; Kang et al., 2015; Poff et al., 2014; Shukla et al., 2014). Вуглець глюкози насамперед спрямовується через гліколіз, пентозофосфатний шлях і ліпогенез, які разом забезпечують проміжні продукти, необхідні для розширення біомаси пухлини (Grabacka et al., 2016; Shukla et al., 2014; Yoshii et al., 2015). Адаптація ракових клітин до нестачі глюкози відбувається через здатність використовувати альтернативні джерела палива, включаючи ацетат, глутамін і аспартат (Jaworski et al., 2016; Sullivan et al., 2015). Наприклад, обмежений доступ до пірувату виявляє здатність ракових клітин перетворювати глутамін в ацетил-КоА шляхом карбоксилування, підтримуючи як енергетичні, так і анаболічні потреби (Yang et al., 2014). Цікавою адаптацією ракових клітин є використання ацетату як палива (Comerford et al., 2014; Jaworski et al., 2016; Mashimo et al., 2014; Wright and Simone, 2016; Yoshii et al., 2015). Ацетат також є субстратом для ліпогенезу, який має вирішальне значення для проліферації пухлинних клітин, і збільшення цього ліпогенного каналу пов'язане з коротшим виживанням пацієнтів і більшим навантаженням на пухлину (Comerford et al., 2014; Mashimo et al., 2014; Yoshii et al. ., 2015).
Неракові клітини легко переміщають джерело енергії з глюкози на кетонові тіла під час депривації глюкози. Ця пластичність може бути більш варіабельною серед типів ракових клітин, але імплантовані пухлини мозку in vivo окислюються [2,4-13C2]-?OHB до того ж ступеня, що й навколишня тканина мозку (De Feyter et al., 2016). Моделі «зворотного ефекту Варбурга» або «метаболізму двох компартментів пухлини» припускають, що ракові клітини індукують вироблення ?OHB у сусідніх фібробластах, забезпечуючи енергетичні потреби пухлинної клітини (Bonuccelli et al., 2010; Martinez-Outschoorn et al., 2012). . У печінці зсув гепатоцитів від кетогенезу до окислення кетонів у клітинах гепатоцелюлярної карциноми (гепатоми) узгоджується з активацією активності BDH1 та SCOT, що спостерігається у двох клітинних лініях гепатоми (Zhang et al., 1989). Дійсно, клітини гепатоми експресують OXCT1 і BDH1 і окислюють кетони, але тільки при голодуванні сироватки (Huang et al., 2016). В якості альтернативи також був запропонований кетогенез пухлинних клітин. Динамічні зрушення в експресії кетогенних генів проявляються під час ракової трансформації епітелію товстої кишки, типу клітин, який зазвичай експресує HMGCS2, і в нещодавній доповіді припускається, що HMGCS2 може бути прогностичним маркером поганого прогнозу при колоректальних і плоскоклітинних карциномах (Camarero et al. 2006; Чен та ін., 2016). Чи потрібна ця асоціація кетогенез, чи підтримка функції HMGCS2, ще потрібно визначити. І навпаки, очевидна продукція ?OHB клітинами меланоми та гліобластоми, стимульована PPAR? агоніст фенофібрату, був пов’язаний із зупинкою росту (Grabacka et al., 2016). Потрібні подальші дослідження, щоб охарактеризувати роль експресії HMGCS2/SCOT, кетогенезу та окислення кетонів у ракових клітинах.
За межами паливного метаболізму кетони нещодавно були залучені в біологію ракових клітин за допомогою сигнального механізму. Аналіз меланоми BRAF-V600E+ вказав на OCT1-залежну індукцію HMGCL у онкогенний BRAF-залежний спосіб (Kang et al., 2015). Збільшення HMGCL корелювало з більш високою концентрацією AcAc в клітинах, що, в свою чергу, посилювало взаємодію BRAFV600E-MEK1, посилюючи передачу сигналів MEK-ERK у петлі прямої подачі, яка стимулює проліферацію та ріст пухлинних клітин. Ці спостереження піднімають інтригуюче питання про перспективний позапечінковий кетогенез, який потім підтримує сигнальний механізм (також див. ?OHB як сигнальний медіатор і Суперечки в позапечінковому кетогенезі). Також важливо враховувати незалежні ефекти AcAc, d-?OHB та l-?OHB на метаболізм раку, і при розгляді HMGCL катаболізм лейцину також може бути порушений.
Ефекти кетогенної дієти (також див. Терапевтичне використання кетогенної дієти та екзогенних кетонових тіл) на моделях ракових тварин різноманітні (De Feyter et al., 2016; Klement et al., 2016; Meidenbauer et al., 2015; Poff et al. ., 2014; Сейфрід та ін., 2011; Шукла та ін., 2014). Хоча епідеміологічні асоціації між ожирінням, раком і кетогенними дієтами обговорюються (Liskiewicz et al., 2016; Wright and Simone, 2016), мета-аналіз із використанням кетогенної дієти на тваринних моделях і в дослідженнях на людях припустив благотворний вплив на виживання, з переваги, проспективно пов’язані з величиною кетозу, часом початку дієти та локалізацією пухлини (Klement et al., 2016; Woolf et al., 2016). Лікування ракових клітин підшлункової залози кетоновими тілами (d-?OHB або AcAc) пригнічує ріст, проліферацію та гліколіз, а кетогенна дієта (81% ккал жирів, 18% білків, 1% вуглеводів) зменшує in vivo масу пухлини, глікемію та збільшення м’язової маси та маси тіла у тварин з імплантованим раком (Shukla et al., 2014). Подібні результати спостерігалися з використанням моделі клітин метастатичної гліобластоми у мишей, які отримували кетонові добавки в раціоні (Poff et al., 2014). І навпаки, кетогенна дієта (91% ккал жиру, 9% білка) підвищувала концентрацію ?OHB в крові та зменшувала глікемію, але не впливала ні на обсяг пухлини, ні на тривалість виживання у щурів з гліомою (De Feyter et al., 2016). Індекс глюкози кетонів був запропонований як клінічний індикатор, який покращує метаболічний контроль при лікуванні раку мозку, спричиненому кетогенною дієтою, у людей та мишей (Meidenbauer et al., 2015). Взявши разом, роль метаболізму кетонових тіл і кетонових тіл у біології раку є надзвичайними, оскільки кожен із них пропонує терапевтичні варіанти, але фундаментальні аспекти залишаються з’ясованими, з чітким впливом, що випливає з матриці змінних, включаючи (i) відмінності між екзогенним кетоном. тіла проти кетогенної дієти, (ii) тип ракової клітини, геномний поліморфізм, ступінь і стадія; і (iii) час і тривалість впливу кетотичного стану.
Кетогенез створюється кетоновими тілами шляхом розщеплення жирних кислот і кетогенних амінокислот. Цей біохімічний процес забезпечує енергією різні органи, зокрема мозок, за умов голодування як відповідь на відсутність глюкози в крові. Кетонові тіла в основному виробляються в мітохондріях клітин печінки. Хоча інші клітини здатні здійснювати кетогенез, вони не настільки ефективні в цьому, як клітини печінки. Оскільки кетогенез відбувається в мітохондріях, його процеси регулюються незалежно. Д-р Алекс Хіменес, округ Колумбія, CCST Insight
Терапевтичне застосування кетогенної дієти та екзогенних кетонових тіл
Застосування кетогенної дієти та кетонових тіл як терапевтичних засобів також виникло в неракових контекстах, включаючи ожиріння та НАЖХП/НАСГ (Browning et al., 2011; Foster et al., 2010; Schugar and Crawford, 2012); серцева недостатність (Huynh, 2016; Kolwicz et al., 2016; Taegtmeyer, 2016); неврологічні та нейродегенеративні захворювання (Martin et al., 2016; McNally and Hartman, 2012; Rho, 2015; Rogawski et al., 2016; Yang and Cheng, 2010; Yao et al., 2011); вроджені порушення метаболізму (Scholl-B�rgi et al, 2015); і виконання вправ (Cox et al., 2016). Ефективність кетогенної дієти була особливо високо оцінена в терапії епілептичних припадків, особливо у пацієнтів, стійких до ліків. Більшість досліджень оцінювали кетогенну дієту у педіатричних пацієнтів і виявляли зниження частоти нападів приблизно на 50% через 3 місяці з покращенням ефективності при окремих синдромах (Wu et al., 2016b). Досвід є більш обмеженим у дорослих епілепсії, але подібне зниження є очевидним, з кращою відповіддю у пацієнтів із симптоматичною генералізованою епілепсією (Nei et al., 2014). Основні протисудомні механізми залишаються неясними, хоча постульовані гіпотези включають зниження утилізації/гліколізу глюкози, перепрограмований транспорт глутамату, непрямий вплив на АТФ-чутливі калієві канали або рецептор аденозину A1, зміну експресії ізоформи натрієвих каналів або вплив на циркулюючий гормон (гормони, що циркулюють в крові). Lambrechts et al., 2016; Lin et al., 2017; Lutas and Yellen, 2013). Залишається незрозумілим, чи протисудомний ефект пов’язаний насамперед із кетоновими тілами, чи через каскадні метаболічні наслідки дієт з низьким вмістом вуглеводів. Тим не менш, кетонові ефіри (див. нижче), здається, підвищують поріг судом на тваринних моделях спровокованих судом (Ciarlone et al., 2016; D'Agostino et al., 2013; Viggiano et al., 2015).
Дієти з низьким вмістом вуглеводів у стилі Аткінса і кетогенні дієти часто вважаються неприємними і можуть спричинити запор, гіперурикемію, гіпокальціємію, гіпомагніємію, призвести до нефролітіазу, кетоацидозу, викликати гіперглікемію та підвищити концентрацію циркулюючого холестерину та вільної жирної кислоти 2001, Kossoff and Hartman, 2012; Kwiterovich et al., 2003; Suzuki et al., 2002). З цих причин довготривала прихильність створює проблеми. Дослідження на гризунах зазвичай використовують характерний розподіл макроелементів (94% ккал жиру, 1% ккал вуглеводів, 5% білка, Bio-Serv F3666), що провокує сильний кетоз. Однак збільшення вмісту білка навіть до 10% ккал істотно зменшує кетоз, а обмеження білка на 5% надає змішані метаболічні та фізіологічні ефекти. Цей дієтичний склад також містить холін, що є ще однією змінною, яка впливає на схильність до пошкоджень печінки і навіть на кетогенез (Garbow et al., 2011; Jornayvaz et al., 2010; Kennedy et al., 2007; Pissios et al., 2013; Schugar та ін., 2013). Вплив тривалого споживання кетогенної дієти на мишах залишається не повністю визначеним, але нещодавні дослідження на мишах показали нормальне виживання та відсутність маркерів ураження печінки у мишей на кетогенних дієтах протягом їхнього життя, хоча метаболізм амінокислот, витрата енергії та передача сигналів інсуліну. були помітно перепрограмовані (Douris et al., 2015).
Механізми, що підвищують кетоз за допомогою механізмів, альтернативних кетогенним дієтам, включають використання попередників кетонових тіл, які вживаються всередину. Введення екзогенних кетонових тіл може створити унікальний фізіологічний стан, який не зустрічається в нормальній фізіології, оскільки концентрації циркулюючої глюкози та інсуліну є відносно нормальними, тоді як клітини можуть знижувати поглинання та використання глюкози. Самі кетонові тіла мають короткий період напіврозпаду, і прийом або вливання солі натрію ?OHB для досягнення терапевтичного кетозу провокує небажане навантаження натрієм. R/S-1,3-бутандіол є нетоксичним діаспиртом, який легко окислюється в печінці з утворенням d/l-?OHB (Desrochers et al., 1992). У різних експериментальних контекстах цю дозу вводили щодня мишам або щурам протягом семи тижнів, що дає циркулюючі концентрації ?OHB до 5 мМ протягом 2 годин після введення, які стабільні протягом щонайменше додаткових 3 годин (D' Агостіно та ін., 2013). Часткове пригнічення споживання їжі спостерігалося у гризунів, які отримували R/S-1,3-бутандіол (Carpenter and Grossman, 1983). Крім того, три хімічно різні ефіри кетонів (KEs), (i) моноефір R-1,3-бутандиолу та d-?OHB (R-3-гідроксибутил R-?OHB); (ii) гліцерил-трис-?OHB; і (iii) диефір ацетоацетату R,S-1,3-бутандиолу також були детально вивчені (Brunengraber, 1997; Clarke et al., 2012a; Clarke et al., 2012b; Desrochers et al., 1995a; Desrochers et al. ., 1995b; Kashiwaya et al., 2010). Невід'ємною перевагою першого є те, що після гідролізу естерази в кишечнику або печінці на моль KE утворюється 2 моль фізіологічного d-?OHB. Безпека, фармакокінетика та переносимість були найбільш детально вивчені у людей, які споживали R-3-гідроксибутил R-?OHB в дозах до 714 мг/кг, що давало циркулюючі концентрації d-?OHB до 6 мМ (Clarke et al., 2012a; Cox et al., 2016; Kemper et al., 2015; Shivva et al., 2016). У гризунів цей KE зменшує споживання калорій і загальний холестерин плазми, стимулює буру жирову тканину та покращує резистентність до інсуліну (Kashiwaya et al., 2010; Kemper et al., 2015; Veech, 2013). Останні результати свідчать про те, що під час фізичних навантажень у тренованих спортсменів прийом R-3-гідроксибутил R-?OHB в скелетних м’язах знижував гліколіз скелетних м’язів і концентрацію лактату в плазмі, посилював внутрішньом’язове окислення триацилгліцерину та зберігав вміст м’язового глікогену, навіть коли одночасно стимулювався секреція інсуліну (стимуляція секреції інсуліну). Кокс та ін., 2016). Потрібна подальша розробка цих інтригуючих результатів, оскільки покращення продуктивності вправ на витривалість переважно було зумовлено стійкою реакцією на КЕ у 2/8 суб’єктів. Тим не менш, ці результати підтверджують класичні дослідження, які вказують на перевагу окислення кетонів перед іншими субстратами (GARLAND et al., 1962; Hasselbaink et al., 2003; Stanley et al., 2003; Valente-Silva et al., 2015), в тому числі під час вправ, і що треновані спортсмени можуть бути більш підготовленими до використання кетонів (Johnson et al., 1969a; Johnson and Walton, 1972; Winder et al., 1974; Winder et al., 1975). Нарешті, механізми, які можуть підтримувати покращення продуктивності вправи після рівного споживання калорій (по-різному розподіленого між макроелементами) та рівного споживання кисню, ще потрібно визначити.
Майбутня перспектива
Після того, як значною мірою стигматизували як шлях переповнення, здатний накопичувати токсичні викиди від спалювання жиру в станах з обмеженим вмістом вуглеводів (парадигма «кетотоксичності»), останні спостереження підтверджують ідею, що метаболізм кетонових тіл виконує оздоровчу роль навіть у станах, насичених вуглеводами, відкриваючи «кетогорметик». � гіпотеза. Хоча легкі харчові та фармакологічні підходи до маніпулювання метаболізмом кетонів роблять його привабливою терапевтичною метою, агресивно поставлені, але розважливі експерименти залишаються як у базових, так і в трансляційних дослідницьких лабораторіях. Незадоволені потреби виникли у сферах визначення ролі метаболізму кетонів при серцевій недостатності, ожирінні, НАЖХП/НАСГ, діабеті 2 типу та раку. Обсяг і вплив «неканонічних» сигнальних ролей кетонових тіл, включаючи регуляцію PTM, які, ймовірно, повертаються і переходять у метаболічні та сигнальні шляхи, потребують більш глибокого дослідження. Нарешті, позапечінковий кетогенез може відкрити інтригуючі паракринні та аутокринні сигнальні механізми та можливості впливати на сумісний метаболізм у нервовій системі та пухлинах для досягнення терапевтичних цілей.
На закінчення, кетонові тіла виробляються печінкою для того, щоб використовуватися як джерело енергії, коли в організмі людини недостатньо глюкози. Кетогенез виникає, коли в крові низький рівень глюкози, особливо після того, як інші клітинні запаси вуглеводів вичерпані. Метою статті вище було обговорити багатовимірну роль кетонових тіл у паливному метаболізмі, передачі сигналів та терапевтиці. Обсяг нашої інформації обмежено хіропрактикою та проблемами здоров’я хребта. Щоб обговорити тему, зверніться до доктора Хіменеса або зв’яжіться з нами за адресою�915-850-0900.
Додаткова тема для обговорення: «Гострий біль у спині».
Біль у спині� є однією з найпоширеніших причин інвалідності та пропущених робочих днів у всьому світі. Біль у спині є другою за поширеністю причиною відвідувань лікаря, переважаючи лише інфекції верхніх дихальних шляхів. Приблизно 80 відсотків населення відчувають біль у спині хоча б раз у житті. Хребет – це складна структура, що складається з кісток, суглобів, зв’язок і м’язів, серед інших м’яких тканин. Травми та/або загострені стани, такі як �грижі диски, може зрештою призвести до симптомів болю в спині. Спортивні травми або травми в автомобільній катастрофі часто є найчастішою причиною болю в спині, однак іноді найпростіші рухи можуть мати хворобливі наслідки. На щастя, альтернативні варіанти лікування, такі як хіропрактика, можуть допомогти полегшити біль у спині за допомогою корекції хребта та ручних маніпуляцій, що в кінцевому підсумку покращує полегшення болю. �
Команда сигнальний шлях фактора 2, пов’язаний з ядерним еритроїдом 2, найбільш відомий як Nrf2, є захисним механізмом, який функціонує як «головний регулятор» антиоксидантної реакції людського організму. Nrf2 визначає рівень окисного стресу в клітинах і запускає захисні антиоксидантні механізми. Хоча активація Nrf2 може мати багато переваг, «надмірна експресія» Nrf2 може мати кілька ризиків.
Здається, що збалансований рівень NRF2 є важливим для запобігання загального розвитку різноманітних захворювань на додаток до загального поліпшення цих проблем зі здоров’ям. Однак NRF2 також може викликати ускладнення. Основною причиною «надекспресії» NRF2 є генетична мутація або триваючий хронічний вплив хімічного або окисного стресу, серед іншого. Нижче ми обговоримо негативні сторони надекспресії Nrf2 і продемонструємо механізми її дії в організмі людини.
рак
Дослідження показали, що миші, які не експресують NRF2, більш схильні до розвитку раку у відповідь на фізичну та хімічну стимуляцію. Однак подібні дослідження показали, що надмірна активація NRF2 або навіть інактивація KEAP1 може призвести до загострення деяких видів раку, особливо якщо ці шляхи були перервані. Надмірна активність NRF2 може виникнути через куріння, де безперервна активація NRF2 вважається причиною раку легенів у курців. Надмірна експресія Nrf2 може призвести до того, що ракові клітини не самознищуються, тоді як періодична активація NRF2 може запобігти індукції токсинів раковими клітинами.
Крім того, оскільки надекспресія NRF2 збільшує антиоксидантну здатність людського організму функціонувати за межами окислювально-відновного гомеостазу, це посилює поділ клітин і створює неприродну схему метилювання ДНК та гістонів. Це в кінцевому підсумку може зробити хіміотерапію та променеву терапію менш ефективними проти раку. Тому обмеження активації NRF2 такими речовинами, як DIM, лютеолін, Zi Cao або саліноміцин, може бути ідеальним для пацієнтів з раком, хоча надмірна активація Nrf2 не повинна розглядатися як єдина причина раку. Дефіцит поживних речовин може вплинути на гени, включаючи NRF2. Це може бути одним із способів того, як недоліки сприяють утворенню пухлин.
Печінка
Надактивація Nrf2 також може вплинути на функцію окремих органів в організмі людини. Надмірна експресія NRF2 може в кінцевому підсумку блокувати вироблення інсуліноподібного фактора росту 1, або IGF-1, у печінці, який є важливим для регенерації печінки.
Серце
У той час як гостра надекспресія Nrf2 може мати свої переваги, постійна надекспресія NRF2 може викликати довгострокові шкідливі ефекти на серце, такі як кардіоміопатія. Експресія NRF2 може бути збільшена за рахунок високого рівня холестерину або активації HO-1. Вважається, що це є причиною того, що хронічний підвищений рівень холестерину може викликати проблеми зі здоров'ям серцево-судинної системи.
Вітіліго
Також було продемонстровано, що надмірна експресія NRF2 пригнічує здатність до репігментації при вітіліго, оскільки вона може перешкоджати дії тирозинази або TYR, яка є важливою для репігментації через меланіногенез. Дослідження показали, що цей процес може бути однією з основних причин того, чому люди з вітіліго, здається, не активують Nrf2 так само ефективно, як люди без вітіліго.
Чому NRF2 може не функціонувати належним чином
Хормесіс
NRF2 має бути активований горметично, щоб мати можливість скористатися його перевагами. Іншими словами, Nrf2 не повинен запускатися щохвилини або щодня, тому було б чудовою ідеєю робити перерви, наприклад, 5 днів на 5 вихідних або через день. NRF2 також повинен досягти певного порогу, щоб запустити його гормональну реакцію, де невеликого стресора може бути недостатньо, щоб його запустити.
DJ-1 Окислення
Білкова дегліказа DJ-1, або просто DJ-1, також званий білком хвороби Паркінсона, або PARK7, є головним регулятором і детектором окислювально-відновного статусу в організмі людини. DJ-1 має важливе значення для регулювання того, як довго NRF2 може виконувати свою функцію та виробляти антиоксидантну відповідь. У разі надмірного окислення DJ-1 клітини зроблять білок DJ-1 менш доступним.
Цей процес спонукає активацію NRF2 занадто швидко закінчуватися, оскільки DJ-1 має першорядне значення для підтримки збалансованих рівнів NRF2 і запобігання їх розпаду в клітині. У випадку, якщо білок DJ-1 не існує або переокислений, експресія NRF2, ймовірно, буде мінімальною, навіть за допомогою DIM або альтернативних активаторів NRF2. Експресія DJ-1 необхідна для відновлення порушеної дії NRF2.
Хронічні захворювання
Якщо у вас є хронічні захворювання, включаючи CIRS, хронічні інфекції/дисбактеріоз/SIBO, або накопичення важких металів, таких як ртуть та/або з кореневих каналів, це може перешкоджати системам NRF2 та фазі детоксикації. Замість того, щоб окислювальний стрес перетворював NRF2 на антиоксидант, NRF2 не спрацьовує, і окислювальний стрес може залишатися в клітині і спричиняти пошкодження, тобто антиоксидантної реакції немає. Це важлива причина, чому багато людей з CIRS мають кілька чутливих і схильні до численних факторів. Деякі люди вважають, що вони можуть мати реакцію herx, однак ця реакція може лише пошкоджувати клітини далі.
Лікування хронічних захворювань, однак, дозволить печінці викидати токсини в жовч, поступово розвиваючи горметичну відповідь активації NRF2. Якщо жовч залишається токсичною і не виводиться з організму людини, вона відновить окислювальний стрес NRF2 і спричинить погіршення самопочуття після того, як вона повторно всмоктується з шлунково-кишкового тракту або шлунково-кишкового тракту. Наприклад, охратоксин А може блокувати NRF2. Крім лікування проблеми, інгібітори гістондеацетилази можуть блокувати окислювальну реакцію ряду факторів, які викликають активацію NRF2, але вони також можуть перешкоджати нормальному запуску NRF2, що в кінцевому підсумку може не виконувати своєї мети.
Порушення регуляції риб’ячого жиру
Холінергічні речовини – це речовини, які підвищують ацетилхолін, або ACh, і холін в мозку через збільшення ACh, особливо при пригніченні розпаду ACh. Пацієнти з CIRS часто мають проблеми з порушенням регуляції рівня ацетилхоліну в організмі людини, особливо в мозку. Риб’ячий жир запускає NRF2, активуючи його захисний антиоксидантний механізм у клітинах.
Люди з хронічними захворюваннями можуть мати проблеми з когнітивним стресом і ексайтотоксичністю ацетилхоліну через накопичення фосфаторганічних речовин, що може призвести до того, що риб’ячий жир викликає запалення в організмі людини. Дефіцит холіну додатково викликає активацію NRF2. Включення холіну у ваш раціон (поліфенолів, яєць тощо) може допомогти посилити ефекти холінергічної дисрегуляції.
Що зменшує NRF2?
Зменшення надмірної експресії NRF2 найкраще для людей з раком, хоча це може бути корисно для багатьох інших проблем зі здоров’ям.
Дієта, харчові добавки та звичайні ліки:
Апігенін (вищі дози)
Бруцея яванська
Каштани
EGCG (високі дози збільшують NRF2)
Пажитник (тригонелін)
Hiba (хінокітіол/?-туяпліцин)
Дієта з високим вмістом солі
Лютеолін (селера, зелений перець, петрушка, лист перілли та ромашковий чай – більш високі дози можуть збільшити NRF2 – 40 мг/кг лютеоліну тричі на тиждень)
Метформін (хронічний прийом)
N-ацетил-L-цистеїн (NAC, блокуючи окислювальну відповідь, особливо у високих дозах)
Кверцетин (вищі дози можуть збільшити NRF2 – 50 мг/кг/день кверцетину)
Саліноміцин (лікарський засіб)
Ретинол (повністю транс-ретиноева кислота)
Вітамін С у поєднанні з кверцетином
Цзи Цао (Пурпурний Громвел має Шиконін/Алканін)
Шляхи та інше:
Бах1
BET
Біоплівки
Брусатол
Камптотецин
DNMT
DPP-23
EZH2
Передача сигналів глюкокортикоїдних рецепторів (дексаметазон і бетаметазон також)
ГСК-3? (регуляторний зворотній зв'язок)
Активація HDAC?
Галофугінон
Гомоцистеїн (ALCAR може змінити цей гомоцистеїн, що викликає низькі рівні NRF2)
IL-24
Keep1
MDA-7
NF?B
Охратоксин А (види Aspergillus і pencicllium)
Білок промієлоцитарної лейкемії
p38
p53
p97
Альфа-рецептор ретиноєвої кислоти
селеніт
SYVN1 (Hrd1)
Інгібування STAT3 (наприклад, криптотаншинон)
Тестостерон (і тестостерону пропіонат, хоча TP інтраназально може збільшити NRF2)
Trecator (етіонамід)
Trx1 (через зменшення Cys151 в Keap1 або Cys506 в області NLS Nrf2)
Тролокс
Вориностат
Дефіцит цинку (погіршує роботу мозку)
Nrf2 Механізм дії
Окислювальний стрес запускається через CUL3, де NRF2 з KEAP1, негативного інгібітора, згодом потрапляє в ядро цих клітин, стимулюючи транскрипцію ARE, перетворюючи сульфіди в дисульфіди та перетворюючи їх на більше антиоксидантних генів, що призводить до активізації антиоксидантів, наприклад як GSH, GPX, GST, SOD тощо. Решту їх можна побачити у списку нижче:
Збільшує AKR
Збільшує АРЕ
Підвищує ATF4
Збільшує Bcl-xL
Підвищує Bcl-2
Підвищує BDNF
Збільшує BRCA1
Збільшує c-червень
Збільшує CAT
Підвищує цГМФ
Підвищує CKIP-1
Підвищує CYP450
Збільшує Cul3
Підвищує ГКЛ
Збільшує GCLC
Збільшує GCLM
Підвищує ГКС
Збільшує GPx
Збільшує GR
Підвищує GSH
Збільшує податок на товари та послуги
Збільшує HIF1
Підвищує HO-1
Підвищує HQO1
Підвищує HSP70
Підвищує рівень ІЛ-4
Підвищує рівень ІЛ-5
Підвищує рівень ІЛ-10
Підвищує рівень ІЛ-13
Збільшує К6
Збільшує К16
Збільшує К17
Збільшує mEH
Збільшує Mrp2-5
Підвищує НАДФН
Підвищує рівень 1
Збільшує NQO1
Підвищує PPAR-альфа
Збільшує Prx
Збільшує р62
Збільшує Sesn2
Збільшує Slco1b2
Збільшує sMafs
Збільшує СОД
Збільшує Trx
Збільшує Txn(d)
Збільшує UGT1(A1/6)
Підвищує VEGF
Зменшує ADAMTS (4/5)
Знижує альфа-SMA
Знижує АЛТ
Зменшує AP1
Знижує АСТ
Зменшує Бах1
Знижує ЦОГ-2
Зменшує DNMT
Знижує FASN
Знижує FGF
Зменшує HDAC
Знижує IFN-?
Знижує IgE
Знижує IGF-1
Знижує рівень IL-1b
Знижує рівень ІЛ-2
Знижує рівень ІЛ-6
Знижує рівень ІЛ-8
Знижує рівень ІЛ-25
Знижує рівень ІЛ-33
Зменшує iNOS
Знижує LT
Зменшує Keap1
Знижує MCP-1
Знижує MIP-2
Знижує MMP-1
Знижує MMP-2
Знижує MMP-3
Знижує MMP-9
Знижує MMP-13
Зменшує NfkB
Зменшує NO
Зменшує SIRT1
Знижує TGF-b1
Знижує TNF-альфа
Знижує Tyr
Зменшує VCAM-1
Закодований з гена NFE2L2, NRF2 або фактор 2, пов’язаний з ядерним еритроїдом 2, є фактором транскрипції в базовій лейциновій блискавці, або bZIP, надсімействі, яка використовує Cap'n'Collar, або структуру CNC.
Він стимулює азотні ферменти, ферменти біотрансформації та транспортери витоку ксенобіотиків.
Це важливий регулятор при індукції генів антиоксидантів і ферментів детоксикації фази II, які захищають клітини від пошкоджень, спричинених окислювальним стресом та електрофільними атаками.
Під час гомеостатичних умов Nrf2 секвеструється в цитозолі шляхом тілесного приєднання N-кінцевого домену Nrf2 або Kelch-подібного ECH-асоційованого білка або Keap1, також званого INrf2 або інгібітором Nrf2, інгібуючи активацію Nrf2.
Він також може контролюватися за допомогою селенопротеїну тіоредоксин редуктази 1 ссавців або TrxR1, який функціонує як негативний регулятор.
Після вразливості до електрофільних стресорів Nrf2 відокремлюється від Keap1, переміщаючись у ядро, де потім гетеродимеризується за допомогою ряду білків-регуляторів транскрипції.
Часті взаємодії включають такі взаємодії органів транскрипції Jun і Fos, які можуть бути членами сімейства білків-активаторів факторів транскрипції.
Після димеризації ці комплекси зв’язуються з компонентами ARE/EpRE, які реагують на антиоксиданти/електрофіли, і активують транскрипцію, як це стосується комплексу Jun-Nrf2, або пригнічують транскрипцію, подібно до комплексу Fos-Nrf2.
Позиціонування ARE, яке запускається або пригнічується, визначить, які гени транскрипційно контролюються цими змінними.
Коли активується ARE:
Активація синтезу антиоксидантів здатна детоксикувати АФК, як каталаза, супероксиддисмутаза або СОД, GSH-пероксидаза, GSH-редуктаза, GSH-трансфераза, НАДФН-хіноноксидоредуктаза або NQO1, цитохром Р450, моноокситіогеназа, система цитохрому Р70 редуктаза і HSPXNUMX.
Активація цієї GSH-синтази забезпечує помітне зростання внутрішньоклітинного рівня GSH, що є досить захисним.
Посилення цього синтезу та ступеня ферментів фази II, таких як UDP-глюкуронозилтрансфераза, N-ацетилтрансферази та сульфотрансферази.
Підвищення регуляції HO-1, який є дійсно захисним рецептором з потенційним зростанням CO, що в поєднанні з NO дозволяє вазодилатацію ішемізованих клітин.
Зменшення перевантаження залізом за рахунок підвищення рівня феритину та білірубіну як ліпофільного антиоксиданту. Обидва білки фази II разом з антиоксидантами здатні фіксувати хронічний окислювальний стрес, а також відновлювати нормальну окислювально-відновну систему.
GSK3? під керівництвом AKT і PI3K фосфорилює Fyn, що призводить до ядерної локалізації Fyn, яка Fyn фосфорилює Nrf2Y568, що призводить до ядерного експорту та деградації Nrf2.
NRF2 також послаблює реакцію TH1/TH17 і збагачує відповідь TH2.
Інгібітори HDAC ініціювали сигнальний шлях Nrf2 і регулювали, що нижній Nrf2 спрямований на HO-1, NQO1 і каталітичну субодиницю глутамат-цистеїн-лігази або GCLC, стримуючи Keap1 і заохочуючи дисоціацію Keap1 від Nrf2, Nrflo і Nrf2. - Є обов'язковими.
Nrf2 включає період напіввиведення приблизно 20 хвилин у базальних умовах.
Зменшення IKK? пул через прив'язку Keap1 зменшує I?B? деградація і може бути невловимим механізмом, за допомогою якого було доведено, що активація Nrf2 інгібує активацію NF?B.
Keap1 не завжди потрібно знижувати, щоб NRF2 працював, наприклад хлорофілін, чорниця, елагова кислота, астаксантин і поліфеноли чаю можуть підвищити NRF2 і KEAP1 на 400 відсотків.
Nrf2 регулює негативно через термін стеароіл-КоА-десатурази, або SCD, і цитрат-ліази, або CL.
Генетика
KEAP1
rs1048290
Алель C – показав значний ризик і захисний ефект проти лікарсько-резистентної епілепсії (DRE)
rs11085735 (я AC)
пов'язаний зі швидкістю зниження функції легенів у LHS
КАРТА
rs242561
Алель T – захисний алель для паркінсонічних розладів – мав міцніше зв’язування NRF2/sMAF і був пов’язаний з вищими рівнями мРНК MAPT у 3 різних областях мозку, включаючи кору мозочка (CRBL), скроневу кору (TCTX), внутрішньодолькову білу речовину (WHMT).
NFE2L2 (NRF2)
rs10183914 (я CT)
Алель T – підвищення рівня білка Nrf2 і відстрочений вік початку хвороби Паркінсона на чотири роки
rs16865105 (я AC)
Алель С – мав більший ризик хвороби Паркінсона
rs1806649 (я CT)
Алель C – був ідентифікований і може мати значення для етіології раку молочної залози.
пов’язані з підвищеним ризиком госпіталізації в періоди високого рівня PM10
rs1962142 (я GG)
Алель T – асоціюється з низьким рівнем експресії NRF2 в цитоплазмі (P = 0.036) і негативною експресією сульфіредоксину (P = 0.042)
Алель – захищений від зниження кровотоку передпліччя (FEV) (об’єм форсованого видиху за одну секунду) по відношенню до куріння сигарети (p = 0.004)
rs2001350 (я TT)
Алель T – захищений від зниження ОФВ (об’єм форсованого видиху за одну секунду) по відношенню до куріння сигарети (р = 0.004)
rs2364722 (я AA)
Алель – захищений від зниження ОФВ (об’єм форсованого видиху за одну секунду) по відношенню до куріння сигарети (р = 0.004)
rs2364723
Алель С – пов’язаний зі значно зниженим ОФВ у японських курців з раком легенів
rs2706110
Алель G – показав значний ризик і захисний ефект проти епілепсії, резистентної до ліків (DRE)
Алелі AA – показали значно знижену експресію KEAP1
Алелі AA – були пов’язані з підвищеним ризиком раку молочної залози (P = 0.011)
rs2886161 (я TT)
Алель Т - пов'язана з хворобою Паркінсона
rs2886162
Алель – був пов’язаний з низькою експресією NRF2 (P = 0.011; OR, 1.988; CI, 1.162×3.400), а генотип AA був пов’язаний з гіршою виживаністю (P = 0.032; HR, 1.687; CI, 1.047×2.748)
rs35652124 (я TT)
Алель – пов’язаний з вищим, пов’язаним із віком на початку хвороби Паркінсона проти алеля G
Алель С – мав збільшення білка NRF2
Алель T – мав менше білка NRF2 і більший ризик серцевих захворювань і кров’яного тиску
rs6706649 (я CC)
Алель C – мав нижчий білок NRF2 і підвищував ризик хвороби Паркінсона
rs6721961 (я GG)
Алель Т – мав нижчий білок NRF2
Алелі ТТ – зв’язок між курінням сигарет у завзятих курців і зниженням якості сперми
Алель TT – асоціюється з підвищеним ризиком раку молочної залози [P = 0.008; АБО 4.656; довірчий інтервал (CI), 1.350×16.063] і алель T були пов’язані з низькою ступенем експресії білка NRF2 (P = 0.0003; OR, 2.420; CI, 1.491×3.926) та негативною експресією SRXN1 (P = 0.047; OR; 1.867; CI = 1.002�3.478)
Алель T – алель також був номінально пов’язаний із 28-денною смертністю, пов’язаною з ALI, після синдрому системної запальної відповіді
Алель T – захищений від зниження ОФВ (об’єм форсованого видиху за одну секунду) по відношенню до куріння сигарети (р = 0.004)
Алель G – пов’язаний з підвищеним ризиком розвитку ALI після серйозної травми у європейців та афроамериканців (відношення шансів, АБО 6.44; 95% довірчий інтервал
Алелі AA – пов’язані з астмою, спричиненою інфекцією
Алелі AA – демонстрували значно знижену експресію гена NRF2 і, як наслідок, підвищений ризик раку легенів, особливо тих, хто коли-небудь курив
Алелі AA – мали значно вищий ризик розвитку ЦД 2 типу (OR 1.77; 95% ДІ 1.26, 2.49; p = 0.011) порівняно з тими, хто має генотипом CC
Алелі AA – сильний зв’язок між відновленням ран і пізньою токсичністю радіації (пов’язаний зі значно більш високим ризиком розвитку пізніх наслідків у афроамериканців з тенденцією у європеоїдної раси)
пов’язані з пероральною терапією естрогенами та ризиком венозної тромбоемболії у жінок у постменопаузі
rs6726395 (я AG)
Алель – захищений від зниження ОФВ1 (об’єм форсованого видиху за одну секунду) по відношенню до куріння сигарети (р = 0.004)
Алель – пов’язаний зі значно зниженим ОФВ1 у японських курців з раком легенів
Алелі GG – мали вищі рівні NRF2 і знижений ризик макулярної дегенерації
Алелі GG – мали вищу виживаність при холангіокарциномі
rs7557529 (я CT)
Алель С - пов'язана з хворобою Паркінсона
Окислювальний стрес та інші стресори можуть викликати пошкодження клітин, що в кінцевому підсумку може призвести до різноманітних проблем зі здоров’ям. Дослідження показали, що активація Nrf2 може сприяти захисному антиоксидантному механізму людського організму, однак дослідники обговорювали, що надмірна експресія Nrf2 може мати величезні ризики для загального здоров’я та самопочуття. Різні типи раку також можуть виникати при надмірній активації Nrf2.
Д-р Алекс Хіменес, округ Колумбія, CCST Insight
Сульфорафан та його вплив на рак, смертність, старіння, мозок і поведінку, хвороби серця тощо
Ізотіоціанати є одними з найважливіших рослинних сполук, які ви можете отримати у своєму раціоні. У цьому відео я роблю для них найповнішу справу, яку коли-небудь робили. Короткий період уваги? Перейдіть до улюбленої теми, натиснувши один із моментів часу нижче. Повна хронологія нижче.
Ключові розділи:
00:01:14 – Рак і смертність
00:19:04 – Старіння
00:26:30 – Мозок і поведінка
00:38:06 – Підсумок
00:40:27 – Доза
Повний графік:
00:00:34 – Представлення сульфорафану, головна тема відео.
00:01:14 – Споживання овочів хрестоцвітних і зниження смертності від усіх причин.
00:02:12 – Ризик раку передміхурової залози.
00:02:23 – Ризик раку сечового міхура.
00:02:34 – Ризик раку легенів у курців.
00:02:48 – Ризик раку молочної залози.
00:03:13 – Гіпотетична: що робити, якщо у вас уже рак? (інтервенційний)
00:03:35 – Імовірний механізм, який керує асоціативними даними раку та смертності.
00:04:38 – Сульфорафан і рак.
00:05:32 – Докази на тваринах, що показують сильний вплив екстракту паростків брокколі на розвиток пухлин сечового міхура у щурів.
00:06:06 – Вплив прямого прийому сульфорафану у пацієнтів з раком передміхурової залози.
00:07:09 – Біоакумуляція метаболітів ізотіоціаната у фактичній тканині молочної залози.
00:08:32 – Пригнічення стовбурових клітин раку молочної залози.
00:08:53 – Урок історії: ще в Стародавньому Римі стверджували, що капустяні гриби мають оздоровчі властивості.
00:09:16 – Здатність сульфорафану посилювати виведення канцерогену (бензолу, акролеїну).
00:09:51 – NRF2 як генетичний перемикач через елементи антиоксидантної реакції.
00:10:10 – Як активація NRF2 посилює виведення канцерогену через глутатіон-S-кон'югати.
00:10:34 – Брюссельська капуста підвищує глутатіон-S-трансферазу і зменшує пошкодження ДНК.
00:11:20 – Напій з проростків брокколі збільшує виведення бензолу на 61%.
00:15:45 – Споживання хрестоцвітних овочів і смертність від серцево-судинних захворювань.
00:16:55 – порошок паростків брокколі покращує рівень ліпідів у крові та загальний ризик серцевих захворювань у діабетиків 2 типу.
00:19:04 – Початок секції старіння.
00:19:21 – Дієта, збагачена сульфорафаном, збільшує тривалість життя жуків від 15 до 30% (за певних умов).
00:20:34 – Важливість слабкого запалення для довголіття.
00:22:05 – Овочі хрестоцвітних і порошок паростків брокколі, здається, зменшують широкий спектр запальних маркерів у людей.
00:23:40 – Підсумок у середині відео: розділи про рак, старіння
00:24:14 – Дослідження на мишах показують, що сульфорафан може покращити адаптивну імунну функцію в літньому віці.
00:25:18 – Сульфорафан покращив ріст волосся у мишачої моделі облисіння. Зображення на 00:26:10.
00:26:30 – Початок розділу «Мозок і поведінка».
00:27:18 – Вплив екстракту паростків брокколі на аутизм.
00:27:48 – Вплив глюкорафаніну на шизофренію.
00:28:17 – Початок обговорення депресії (правдоподібний механізм та дослідження).
00:31:21 – Дослідження на мишах з використанням 10 різних моделей депресії, викликаної стресом, показало, що сульфорафан так само ефективний, як і флуоксетин (прозак).
00:32:00 – Дослідження показує, що пряме вживання глюкорафаніну мишами так само ефективне для запобігання депресії через модель стресу соціальної поразки.
00:33:01 – Початок відділу нейродегенерації.
00:33:30 – Сульфорафан і хвороба Альцгеймера.
00:33:44 – Сульфорафан і хвороба Паркінсона.
00:33:51 – Сульфорафан і хвороба Хантінгтона.
00:34:13 – Сульфорафан збільшує кількість білків теплового шоку.
00:34:43 – Початок секції черепно-мозкової травми.
00:35:01 – Сульфорафан, введений відразу після ЧМТ, покращує пам’ять (дослідження на мишах).
00:35:55 – Сульфорафан і нейрональна пластичність.
00:36:32 – Сульфорафан покращує навчання на моделі діабету ІІ типу у мишей.
00:37:19 – Сульфорафанова і м’язова дистрофія Дюшенна.
00:37:44 – Інгібування міостатину в клітинах-супутниках м’язів (in vitro).
00:38:06 – Пізнє відео: смертність і рак, пошкодження ДНК, окислювальний стрес і запалення, виділення бензолу, серцево-судинні захворювання, діабет ІІ типу, вплив на мозок (депресія, аутизм, шизофренія, нейродегенерація), шлях NRF2.
00:40:27 – Думки щодо визначення дози паростків брокколі або сульфорафану.
00:41:01 – Анекдоти про проростання в домашніх умовах.
00:43:14 – Про температуру приготування та активність сульфорафану.
00:43:45 – Перетворення сульфорафану з глюкорафаніну кишковими бактеріями.
00:44:24 – Добавки працюють краще в поєднанні з активною мирозиназою з овочів.
00:44:56 – Техніка приготування та овочі хрестоцвітних.
00:46:06 – Ізотіоціанати як зоб.
Згідно з дослідженнями, Nrf2 є основним фактором транскрипції, який активує захисні антиоксидантні механізми клітин для детоксикації організму людини. Однак надмірна експресія Nrf2 може викликати проблеми зі здоров’ям. Обсяг нашої інформації обмежено хіропрактикою та проблемами здоров’я хребта. Щоб обговорити тему, зверніться до доктора Хіменеса або зв’яжіться з нами за адресою�915-850-0900.
Куратор доктор Алекс Хіменес
Додаткова тема для обговорення: «Гострий біль у спині».
Біль у спині� є однією з найпоширеніших причин інвалідності та пропущених робочих днів у всьому світі. Біль у спині є другою за поширеністю причиною відвідувань лікаря, переважаючи лише інфекції верхніх дихальних шляхів. Приблизно 80 відсотків населення відчувають біль у спині хоча б раз у житті. Хребет – це складна структура, що складається з кісток, суглобів, зв’язок і м’язів, серед інших м’яких тканин. Травми та/або загострені стани, такі як �грижі диски, може зрештою призвести до симптомів болю в спині. Спортивні травми або травми в автомобільній катастрофі часто є найчастішою причиною болю в спині, однак іноді найпростіші рухи можуть мати хворобливі наслідки. На щастя, альтернативні варіанти лікування, такі як хіропрактика, можуть допомогти полегшити біль у спині за допомогою корекції хребта та ручних маніпуляцій, що в кінцевому підсумку покращує полегшення болю.
Багато сучасних досліджень раку дозволили медичним працівникам зрозуміти, як організм детоксикує. Аналізуючи активізовані гени в пухлинних клітинах, дослідники виявили сигнальний шлях фактора 2, пов’язаний з ядерним еритроїдом 2, найбільш відомий як Nrf2. NRF2 є важливим фактором транскрипції, який активує людський організм захисні антиоксидантні механізми для того, щоб регулювати окислення як зовнішніх, так і внутрішніх факторів, щоб запобігти підвищеним рівням окисного стресу.
Принципи Nrf2
NRF2 має важливе значення для підтримки загального здоров’я та самопочуття, оскільки він служить головній меті регулювання того, як ми керуємо всім, з чим ми стикаємося щодня, і не хворіємо. Активація NRF2 відіграє важливу роль у системі детоксикації фази II. Фаза II детоксикації забирає ліпофільні або «жиророзчинні вільні радикали та перетворює їх у гідрофільні або водорозчинні речовини для виведення, одночасно інактивуючи виключно реакційноздатні метаболіти та хімічні речовини, як наслідок фази I.
Активація NRF2 зменшує загальне окислення та запалення людського організму за рахунок горметичного ефекту. Щоб запустити NRF2, має виникнути запальна реакція через окислення, щоб клітини виробляли адаптивну відповідь і створювали антиоксиданти, такі як глутатіон. Щоб порушити принцип Nrf2, по суті, окислювальний стрес активує NRF2, який потім активує антиоксидантну відповідь в організмі людини. NRF2 функціонує, щоб збалансувати окислювально-відновну передачу сигналів або рівновагу рівнів окислювача та антиоксиданту в клітині.
Чудову ілюстрацію того, як цей процес функціонує, можна продемонструвати за допомогою вправ. Під час кожного тренування м’яз адаптується, щоб він міг прийняти ще одне тренування. Якщо NRF2 стає недостатньо або надмірно експресованим через хронічні інфекції або підвищений вплив токсинів, що може спостерігатися у пацієнтів із синдромом хронічної запальної відповіді або CIRS, проблеми зі здоров’ям можуть погіршитися – після активації NRF2. Перш за все, якщо DJ-1 стане надмірно окисленим, активація NRF2 закінчиться занадто швидко.
Ефекти активації NRF2
Активація NRF2 сильно виражена в легенях, печінці та нирках. Фактор 2, пов’язаний з ядерним еритроїдом 2, або NRF2, найчастіше функціонує, протидіючи підвищеному рівню окислення в організмі людини, що може призвести до окисного стресу. Активація Nrf2 може допомогти лікувати різноманітні проблеми зі здоров’ям, однак надмірна активація Nrf2 може погіршити різні проблеми, які продемонстровані нижче.
Періодична активація Nrf2 може допомогти:
Старіння (тобто довголіття)
Аутоімунітет і загальне запалення (наприклад, артрит, аутизм)
Рак і хіміопротекція (тобто вплив ЕМП)
Депресія і тривога (тобто посттравматичний стресовий стресовий синдром)
Вплив наркотиків (алкоголь, НПЗП)
Вправи та Витривалість
Захворювання кишечника (наприклад, SIBO, дисбактеріоз, виразковий коліт)
Захворювання нирок (наприклад, гостре ураження нирок, хронічне захворювання нирок, вовчаковий нефрит)
Захворювання печінки (наприклад, алкогольна хвороба печінки, гострий гепатит, неалкогольна жирова хвороба печінки, неалкогольний стеатогепатит, цироз)
Захворювання легенів (наприклад, астма, фіброз)
Метаболічні та судинні захворювання (наприклад, атеросклероз, гіпертонія, інсульт, цукровий діабет)
Нейродегенерація (тобто хвороба Альцгеймера, Паркінсона, Хантінгтона та БАС)
Біль (тобто нейропатія)
Захворювання шкіри (наприклад, псоріаз, захист від ультрафіолету/сонця)
Вплив токсинів (миш'як, азбест, кадмій, фтор, гліфосат, ртуть, сепсис, дим)
Зір (тобто яскраве світло, чутливість, катаракта, дистрофія рогівки)
Трансплантація серця (хоча відкритий NRF2 може бути поганим, NRF2 може допомогти з відновленням)
Гепатит С
Нефрит (важкі випадки)
Вітіліго
Крім того, NRF2 може допомогти конкретні харчові добавки, ліки та ліки працювати. Багато натуральних добавок також можуть сприяти запуску NRF2. Завдяки поточним дослідженням дослідники продемонстрували, що велика кількість сполук, які колись вважалися антиоксидантами, дійсно є прооксидантами. Це тому, що майже всім їм для функціонування потрібен NRF2, навіть такі добавки, як куркумін і риб’ячий жир. Наприклад, було показано, що какао викликає антиоксидантну дію у мишей, які мають ген NRF2.
Способи активації NRF2
У разі нейродегенеративних захворювань, таких як хвороба Альцгеймера, хвороба Паркінсона, інсульт або навіть аутоімунні захворювання, мабуть, краще підвищити рівень Nrf2, але гормональним шляхом. Змішування активаторів NRF2 також може мати адитивний або синергетичний ефект, оскільки іноді це може бути дозозалежним. Нижче наведено основні способи збільшення виразу Nrf2:
HIST (Вправа) + CoQ10 + Sun (вони дуже добре поєднуються)
Паростки брокколі + LLLT на моїй голові та кишечнику
Бутират + Супер кава + Ранкове сонце
Акупунктура (це альтернативний метод, також може використовуватися лазерна акупунктура)
Пост
Каннабідіола (КБР)
Левова грива + мелатонін
Альфа-ліпоєва кислота + DIM
Полин гіркий
PPAR-гамма-активація
Наведений нижче вичерпний список, що містить понад 350 інших способів активації Nrf2 за допомогою дієти, способу життя та пристроїв, пробіотиків, добавок, трав і масел, гормонів і нейромедіаторів, ліків / ліків і хімікатів, шляхів / факторів транскрипції, а також інших способів, є лише короткий посібник щодо того, що може викликати Nrf2. Для стислості в цій статті ми виключили понад 500 інших продуктів харчування, харчових добавок і сполук, які можуть допомогти активувати Nrf2. Нижче наведено:
Дієта:
Acai Berries
Алкоголь (Краще червоне вино, особливо якщо в ньому є пробка, оскільки протокатехічний альдегід з пробок також може активувати NRF2. Загалом, алкоголь не рекомендується, хоча гострий прийом збільшує NRF2. Хронічний прийом може знизити NRF2.
Водорості (ламінарія)
яблука
Чорний чай
Бразильський горіх
Паростки брокколі (та інші ізотіоціанати, сульфорафан, а також овочі хрестоцвітих, такі як бок-чой, які мають D3T)
Чорниця (0.6-10 г/добу)
Морква (фалькаринон)
Кайєнський перець (капсаїцин)
Селера (бутилфталід)
чага (бетулін)
Ромашковий чай
Чіа
Китайська картопля
Чорноплідна горобина (аронія)
Шоколад (темний або какао)
Кориця
Кава (наприклад, хлорогенова кислота, Cafestol і Kahweol)
Фізичні вправи (гострі вправи, такі як HIST або HIIT, здається, є більш корисними для індукції NRF2, тоді як тривалі вправи не викликають NRF2, але підвищують рівень глутатіону)
Дієта з високим вмістом жиру (дієта)
Висока температура (сауна)
Вдихання водню та воднева вода
Гіпербарична киснева терапія
Інфрачервона терапія (наприклад, Joovv)
Вітамін С внутрішньовенно
Багатий жирами раціон
озон
Куріння (не рекомендується – гостре куріння збільшує NRF2, хронічне куріння знижує NRF2. Якщо ви вирішите курити, святий базилік може допомогти захистити від зниження регуляції NRF2)
Сонце (UVB та інфрачервоне)
Пробіотики:
Bacillus subtilis (fmbJ)
Clostridium butyricum (MIYAIRI 588)
Lactobacillus brevis
Lactobacillus casei (SC4 і 114001)
Lactobacillus collinoides
Lactobacillus gasseri (OLL2809, L13-Ia і SBT2055)
Lactobacillus helveticus (NS8)
Lactobacillus paracasei (NTU 101)
Lactobacillus plantarum (C88, CAI6, FC225, SC4)
Lactobacillus rhamnosus (GG)
Добавки, трави та масла:
Ацетил-L-карнітин (ALCAR) і карнітин
Аліцин
Альфа-ліпоєва кислота
Аментофлавоне
Андрографіс волотистому
Агматин
Апігенин
Аргінін
Артишок (ціанропікрин)
Ашваганда
Астрагал
Bacopa
біфштекс (ізогемакетон)
берберин
Бета-каріофіллен
Біденс Пілос
Олія насіння чорного кмину (тимохінон)
Boswellia
Бутеїн
Бутірат
Каннабідіола (КБР)
Каротиноїди (наприклад, бета-каротин [синергія з лікопіном – 2 × 15 мг/добу лікопіну], фукоксантин, зеаксантин, астаксантин і лютеїн)
Читрак
Хлорела
Хлорофіл
Хризантема Завадська
коричний
Росичка звичайна
Мідь
Коптіс
CoQ10
Куркумін
Даміана
Ден Шен/Червоний шавлія (Мілтірон)
DIM
Діосцин
Донг Лінг Цао
Донг Куай (жіночий женьшень)
Ecklonia Cava
EGCG
Оман / Інула
Кора Eucommia
Ферулінова кислота
Фісетин
Риб’ячий жир (DHA/EPA – 3 × 1 г/день риб’ячого жиру, що містить 1098 мг EPA і 549 мг DHA)
Галангал
Гастродін (Тянь Ма)
Гентіана
герань
Гінкго білоба (гінкголід B)
Склянка
Готу Кола
Екстракт кісточок винограду
Волохатий Агрімоній
Харітакі (Тріфала)
Глід
Гелікрисум
Хна (Juglone)
Гібіскус
Гігенамін
Святий базилік/Тулсі (урсолова кислота)
Хміль
Horny Goat Weed (Ікаріїн/Ікарізид)
Індиго Naturalis
Залізо (не рекомендується, якщо немає необхідності)
I3C
Сльози Йова
Moringa Oleifera (наприклад, Кемпферол)
Інчінкото (комбінація Чжи Цзи та Полину)
Корінь Кудзу
Корінь солодки
Корінь Ліндери
Лютеолін (високі дози для активації, менші дози інгібують NRF2 при раку)
Магнолія
Манджистха
Maximowiczianum (ацерогенін А)
Мексиканська арніка
Расторопша плямиста
MitoQ
Му Сян
Mucuna Pruriens
Нікотинамід і НАД+
Женьшень
Пасифлора (наприклад, Chrysin, але хіризин також може зменшувати NRF2 через порушення регуляції передачі сигналів PI3K/Akt)
Пау д'арко (лапачо)
Флоретин
Piceatannol
PQQ
Процианидин
Птеростільбен
Пуерарія
Кверцетин (тільки високі дози, менші дози інгібують NRF2)
Цян Хо
Червоний конюшина
Ресвератрол (піцеїд та інші фітоестрогени, по суті, спориш)
Плоди шипшини
Рожеве дерево
Рутин
Запалене дерево
Сарсапарелі
Saururus chinensis
SC-E1 (гіпс, жасмин, солодка, кудзу і квітка повітряної кулі)
Лимонник
Самолікування (прунела)
Тюбетейка (Байкалін і Вогонін)
Вівці Щавель
Сі Ву Тан
Сідерит
Spikenard (Аралія)
спіруліна
Іоанна
Сульфорафан
Сазерландія
Тао Хун Сі Ву
Таурин
Виноградний Бог Грому (Тріптолід)
Токофероли (такі як вітамін Е або ліналоол)
Трібулус Р
Ту Сі Цзи
TUDCA
Вітамін А (хоча інші ретиноїди інгібують NRF2)
Вітамін С (тільки висока доза, низька доза пригнічує NRF2)
Вітекс / Цнотливе дерево
Біла півонія (Paeoniflorin з Paeonia lactiflora)
Полин (гіспідулін і артемізинін)
Сяо Яо Ван (вільний і легкий мандрівник)
Єрба Санта (Eriodictyol)
Юань Чжи (Тенуігенін)
Цзи Цао (зменшить NRF2 при раку)
цинк
Зизифус зизифус
Гормони та нейромедіатори:
адипонектин
Адропін
Естроген (але може знижувати NRF2 в тканині молочної залози)
Мелатонін
Прогестерон
Хінолінова кислота (як захисна реакція для запобігання ексайтотоксичності)
серотонін
Гормони щитовидної залози, такі як Т3 (можуть збільшити NRF2 у здорових клітинах, але зменшити його при раку)
Вітамін D
Наркотики/ліки та хімічні речовини:
Ацетамінофен
Ацеталозаміда
Амлодипін
Ауранофін
Бардоксолон метил (BARD)
Бензидазол
ВНА
CDDO-імідазолід
Цефтриаксон (і бета-лактамні антибіотики)
Cialis
Дексаметазон
Диприван (пропофол)
Еріодіктіол
Exendin-4
Езетимібе
Фторид
Фумарат
HNE (окислений)
Ідазоксан
Неорганічний миш'як і арсеніт натрію
JQ1 (може також інгібувати NRF2, невідомо)
Letairis
Мелфалан
Метазоламід
Метиленовий синій
Ніфедипін
НПЗЗ
Ольтіпраз
ІПП (такі як омепразол і лансопразол)
Протандім – чудові результати in vivo, але слабкі/неіснуючі при активації NRF2 у людей
Пробукол
рапаміцин
Reserpine
Рутеній
Ситаксетан
Статини (такі як Ліпітор і Симвастатин)
Тамоксифен
Тан Ло Нін
tBHQ
Текфідера (диметилфумарат)
THC (не такий сильний, як CBD)
Теофілін
Коротко
Урсодезоксихолева кислота (УДХК)
верапаміл
Віагра
4-Ацетоксифенол
Шляхи/Фактори транскрипції:
?7 активація нХР
AMPK
Білірубін
CDK20
ЦКІП-1
CYP2E1
EAATs
Ганкирін
Гремлін
GJA1
Н-феритинфероксидаза
Інгібітори HDAC (такі як вальпроєва кислота і TSA, але можуть викликати нестабільність NRF2)
Білки теплового шоку
IL-17
IL-22
Клото
let-7 (збиває РНК mBach1)
MAPK
Приймачі Майкла (більшість)
МіР-141
МіР-153
miR-155 (збиває також РНК mBach1)
miR-7 (в мозку, допомагає при раку та шизофренії)
Виїмка 1
Окислювальний стрес (наприклад, ROS, RNS, H2O2) та електрофіли
PGC-1?
PKC-дельта
PPAR-гамма (синергетичні ефекти)
Пригнічення рецепторів сигма-1
SIRT1 (збільшує NRF2 в мозку та легенях, але може зменшити його в цілому)
SIRT2
SIRT6 (у печінці та мозку)
SRXN1
Інгібування TrxR1 (також ослаблення або виснаження)
Протопорфірин цинку
4-HHE
Інший:
Анкафлавін
Азбест
Авіцини
Bacillus amyloliquefaciens (використовується в сільському господарстві)
Чадний газ
Дафнетін
Виснаження глутатіону (можливе виснаження на 80% - 90%)
Гімнастер корейський
Гепатит С
герпес (ВПГ)
Індійський ясен
Корінь індиговади
ізосаліпурпозид
Ізорхаментин
Монасцин
Омавелоксолон (сильний, він же RTA-408)
PDTC
Дефіцит селену (дефіцит селену може збільшити NRF2)
Модрина сибірська
Софорафлаванон Г
Tadehagi triquetrum
Toona sinensis (7-DGD)
Квітка труба
63171 і 63179 (сильні)
Сигнальний шлях фактора 2, пов’язаний з ядерним еритроїдом 2, найбільш відомий під акронімом Nrf2, є фактором транскрипції, який відіграє головну роль у регуляції захисних антиоксидантних механізмів людського організму, зокрема, для контролю окисного стресу. Хоча підвищений рівень окисного стресу може активувати Nrf2, його вплив значно посилюється завдяки наявності специфічних сполук. Деякі продукти харчування та добавки сприяють активації Nrf2 в організмі людини, у тому числі ізотіоціанат сульфорафан з паростків брокколі. Д-р Алекс Хіменес, округ Колумбія, CCST Insight
Сульфорафан та його вплив на рак, смертність, старіння, мозок і поведінку, хвороби серця тощо
Ізотіоціанати є одними з найважливіших рослинних сполук, які ви можете отримати у своєму раціоні. У цьому відео я роблю для них найповнішу справу, яку коли-небудь робили. Короткий період уваги? Перейдіть до улюбленої теми, натиснувши один із моментів часу нижче. Повна хронологія нижче.
Ключові розділи:
00:01:14 – Рак і смертність
00:19:04 – Старіння
00:26:30 – Мозок і поведінка
00:38:06 – Підсумок
00:40:27 – Доза
Повний графік:
00:00:34 – Представлення сульфорафану, головна тема відео.
00:01:14 – Споживання овочів хрестоцвітних і зниження смертності від усіх причин.
00:02:12 – Ризик раку передміхурової залози.
00:02:23 – Ризик раку сечового міхура.
00:02:34 – Ризик раку легенів у курців.
00:02:48 – Ризик раку молочної залози.
00:03:13 – Гіпотетична: що робити, якщо у вас уже рак? (інтервенційний)
00:03:35 – Імовірний механізм, який керує асоціативними даними раку та смертності.
00:04:38 – Сульфорафан і рак.
00:05:32 – Докази на тваринах, що показують сильний вплив екстракту паростків брокколі на розвиток пухлин сечового міхура у щурів.
00:06:06 – Вплив прямого прийому сульфорафану у пацієнтів з раком передміхурової залози.
00:07:09 – Біоакумуляція метаболітів ізотіоціаната у фактичній тканині молочної залози.
00:08:32 – Пригнічення стовбурових клітин раку молочної залози.
00:08:53 – Урок історії: ще в Стародавньому Римі стверджували, що капустяні гриби мають оздоровчі властивості.
00:09:16 – Здатність сульфорафану посилювати виведення канцерогену (бензолу, акролеїну).
00:09:51 – NRF2 як генетичний перемикач через елементи антиоксидантної реакції.
00:10:10 – Як активація NRF2 посилює виведення канцерогену через глутатіон-S-кон'югати.
00:10:34 – Брюссельська капуста підвищує глутатіон-S-трансферазу і зменшує пошкодження ДНК.
00:11:20 – Напій з проростків брокколі збільшує виведення бензолу на 61%.
00:15:45 – Споживання хрестоцвітних овочів і смертність від серцево-судинних захворювань.
00:16:55 – порошок паростків брокколі покращує рівень ліпідів у крові та загальний ризик серцевих захворювань у діабетиків 2 типу.
00:19:04 – Початок секції старіння.
00:19:21 – Дієта, збагачена сульфорафаном, збільшує тривалість життя жуків від 15 до 30% (за певних умов).
00:20:34 – Важливість слабкого запалення для довголіття.
00:22:05 – Овочі хрестоцвітних і порошок паростків брокколі, здається, зменшують широкий спектр запальних маркерів у людей.
00:23:40 – Підсумок у середині відео: розділи про рак, старіння
00:24:14 – Дослідження на мишах показують, що сульфорафан може покращити адаптивну імунну функцію в літньому віці.
00:25:18 – Сульфорафан покращив ріст волосся у мишачої моделі облисіння. Зображення на 00:26:10.
00:26:30 – Початок розділу «Мозок і поведінка».
00:27:18 – Вплив екстракту паростків брокколі на аутизм.
00:27:48 – Вплив глюкорафаніну на шизофренію.
00:28:17 – Початок обговорення депресії (правдоподібний механізм та дослідження).
00:31:21 – Дослідження на мишах з використанням 10 різних моделей депресії, викликаної стресом, показало, що сульфорафан так само ефективний, як і флуоксетин (прозак).
00:32:00 – Дослідження показує, що пряме вживання глюкорафаніну мишами так само ефективне для запобігання депресії через модель стресу соціальної поразки.
00:33:01 – Початок відділу нейродегенерації.
00:33:30 – Сульфорафан і хвороба Альцгеймера.
00:33:44 – Сульфорафан і хвороба Паркінсона.
00:33:51 – Сульфорафан і хвороба Хантінгтона.
00:34:13 – Сульфорафан збільшує кількість білків теплового шоку.
00:34:43 – Початок секції черепно-мозкової травми.
00:35:01 – Сульфорафан, введений відразу після ЧМТ, покращує пам’ять (дослідження на мишах).
00:35:55 – Сульфорафан і нейрональна пластичність.
00:36:32 – Сульфорафан покращує навчання на моделі діабету ІІ типу у мишей.
00:37:19 – Сульфорафанова і м’язова дистрофія Дюшенна.
00:37:44 – Інгібування міостатину в клітинах-супутниках м’язів (in vitro).
00:38:06 – Пізнє відео: смертність і рак, пошкодження ДНК, окислювальний стрес і запалення, виділення бензолу, серцево-судинні захворювання, діабет ІІ типу, вплив на мозок (депресія, аутизм, шизофренія, нейродегенерація), шлях NRF2.
00:40:27 – Думки щодо визначення дози паростків брокколі або сульфорафану.
00:41:01 – Анекдоти про проростання в домашніх умовах.
00:43:14 – Про температуру приготування та активність сульфорафану.
00:43:45 – Перетворення сульфорафану з глюкорафаніну кишковими бактеріями.
00:44:24 – Добавки працюють краще в поєднанні з активною мирозиназою з овочів.
00:44:56 – Техніка приготування та овочі хрестоцвітних.
00:46:06 – Ізотіоціанати як зоб.
Згідно з багатьма поточними дослідженнями, сигнальний шлях фактора 2, пов’язаний з ядерним еритроїдом 2, найбільш відомий як Nrf2, є основним фактором транскрипції, який активує захисні антиоксидантні механізми клітин для детоксикації людського організму як від зовнішніх, так і внутрішніх факторів і запобігання підвищенню рівень окисного стресу. Обсяг нашої інформації обмежено хіропрактикою та проблемами здоров’я хребта. Щоб обговорити тему, зверніться до доктора Хіменеса або зв’яжіться з нами за адресою�915-850-0900.
Куратор доктор Алекс Хіменес
Додаткова тема для обговорення: «Гострий біль у спині».
Біль у спині� є однією з найпоширеніших причин інвалідності та пропущених робочих днів у всьому світі. Біль у спині є другою за поширеністю причиною відвідувань лікаря, переважаючи лише інфекції верхніх дихальних шляхів. Приблизно 80 відсотків населення відчувають біль у спині хоча б раз у житті. Хребет – це складна структура, що складається з кісток, суглобів, зв’язок і м’язів, серед інших м’яких тканин. Травми та/або загострені стани, такі як �грижі диски, може зрештою призвести до симптомів болю в спині. Спортивні травми або травми в автомобільній катастрофі часто є найчастішою причиною болю в спині, однак іноді найпростіші рухи можуть мати хворобливі наслідки. На щастя, альтернативні варіанти лікування, такі як хіропрактика, можуть допомогти полегшити біль у спині за допомогою корекції хребта та ручних маніпуляцій, що в кінцевому підсумку покращує полегшення болю. �
Окислювальний стрес є основним фактором розвитку різноманітних проблем зі здоров’ям, включаючи рак, хвороби серця, діабет, прискорене старіння та нейродегенерацію. Продукти, трави та добавки, багаті антиоксидантами, можна використовувати для захисту людського організму від високого рівня окисного стресу. Останні дослідження показали, що Шлях гена Nrf2 може допомогти посилити дію антиоксидантів. The переваги Nrf2 описані нижче.
Захищає організм від токсинів
NRF2 є внутрішньою речовиною, яка може захищати клітини від шкідливих внутрішніх і зовнішніх сполук. NRF2 може допомогти збагатити реакцію організму людини на ліки/ліки та токсини, покращуючи виробництво білків, які допомагають виводити з клітини сполуки, відомі як білки, пов’язані з множинною лікарською стійкістю, або MRP. Наприклад, NRF2 запускається при вдихання сигаретного диму для детоксикації легенів.
Крім того, легеням важливо захищати себе від алергенів, вірусних захворювань, бактеріальних ендотоксинів, гіпероксії та різних забруднювачів навколишнього середовища. Однак постійний тригер Nrf2 може знизити рівень речовини, відомої як глутатіон, в організмі людини. NRF2 також може захищати печінку від токсичності і може захистити печінку від гепатотоксичності миш’яку. Крім того, NRF2 захищає печінку та мозок від вживання алкоголю. Наприклад, Nrf2 може захистити від токсичності ацетамінофену.
Бореться із запаленням та окислювальним стресом
Активація NRF2 може допомогти боротися із запаленням, зменшуючи кількість запальних цитокінів, наприклад тих, що присутні при псоріазі. NRF2 також може зменшити запалення, пов’язане з різними проблемами зі здоров’ям, такими як артрит і фіброз печінки, нирок і легенів. NRF2 також може допомогти контролювати алергію, знижуючи рівень цитокінів Th1/Th17 і підвищуючи рівень цитокінів TH2. Це може бути корисно при таких захворюваннях, як астма.
NRF2 додатково захищає клітини від пошкоджень синім світлом і від UVA/UVB, які виявляються в сонячному світлі. Дефіцит Nrf2 може значно полегшити отримання сонячних опіків. Однією з причин цього є те, що NRF2 здатний регулювати колаген у відповідь на УФ-випромінювання. Удосконалені кінцеві продукти глікації, або AGE, сприяють розвитку багатьох проблем зі здоров’ям, включаючи діабет та нейродегенеративні захворювання. NRF2 може зменшити окислювальний стрес AGE в організмі. NRF2 також може захищати організм людини від вищого рівня теплового стресу.
Підвищує продуктивність мітохондрій і вправ
NRF2 є мітохондріальним бустером. Активація NRF2 сприяє підвищенню енергії АТФ для мітохондрій, на додаток до посиленого використання кисню, або цитрату, і жиру. Без NRF2 мітохондрії просто мали б здатність функціонувати з цукром або глюкозою, а не жиром. NRF2 також необхідний для розвитку мітохондрій за допомогою процесу, відомого як біогенез. Активація NRF2 є життєво важливою, щоб «скористатися» перевагами вправ.
Завдяки активності Nrf2 фізичні вправи підвищують функцію мітохондрій, де цей результат може бути посилений за допомогою CoQ10, кордицепсу та обмеження калорійності. Помірні фізичні навантаження або гострі фізичні навантаження викликають біогенез мітохондрій і підвищений синтез супероксиддисмутази, або СОД, і гем-оксигенази-1, або HO-1, через активацію NRF2. Альфа-ліпоєва кислота, або ALA, і Dan Shen можуть посилити опосередкований NRF2 мітохондріальний біогенез. Крім того, �NRF2 також може покращити толерантність до фізичних навантажень, коли видалення NRF2 робить вправи шкідливими.
Захищає від гіпоксії
NRF2 також допомагає захистити організм людини від втрати/виснаження клітинного кисню, проблеми зі здоров’ям, яка називається гіпоксією. Люди з CIRS мають знижений рівень кисню, оскільки їх NRF2 обструкція, що призводить до зниження рівнів як VEGF, HIF1, так і HO-1. Як правило, у здорових людей з гіпоксією, miR-101, який необхідний для створення стовбурових клітин, надмірно експресується і збільшує кількість NRF2/HO-1 і VEGF/eNOS, таким чином запобігаючи пошкодженню мозку, але, здається, цього не відбувається. в CIRS.
Гіпоксія, що характеризується низьким HIF1, при CIRS також може призвести до негерметичності гематоенцефалічного бар’єру через дисбаланс NRF2. Салідрозид, що знаходиться в родіолі, функціонує на активацію NRF2 і допомагає при гіпоксії, підвищуючи рівні VEGF і HIF1 в організмі людини. NRF2 також може в кінцевому підсумку захистити від накопичення лактату в серці. Активація NRF2 також може зупинити спричинену гіпоксією висотну хворобу руху, або AMS.
Уповільнює старіння
Деякі сполуки, які можуть бути смертельними у великих кількостях, можуть збільшити довговічність у досить незначних кількостях через ксеногормез через NRF2, PPAR-гамма та FOXO. Дуже мала кількість токсинів підвищує здатність клітини ставати краще підготовленими для наступного контакту з токсином, однак це не є схваленням споживання отруйних хімікатів.
Гарною ілюстрацією цього процесу є обмеження калорійності. NRF2 може покращити тривалість життя клітин, підвищуючи їх рівень мітохондрій і антиоксидантів, а також знижуючи здатність клітин до смерті. NRF2 зменшується зі старінням, оскільки NRF2 запобігає загибелі стовбурових клітин і допомагає їм регенерувати. NRF2 відіграє роль у посиленні загоєння ран.
Зміцнює судинну систему
Якщо виробляти сульфорафан правильно, активація NRF2 може захистити від серцевих захворювань, таких як високий кров’яний тиск, гіпертонія, затвердіння артерій або атеросклероз. NRF2 може посилити розслаблюючу активність ацетилхоліну або ACh на судинну систему, одночасно зменшуючи стрес, спричинений холестерином. Активація Nrf2 може зміцнити серце, однак надмірна активація Nrf2 може підвищити ймовірність серцево-судинних захворювань.
Статини можуть запобігти або призвести до серцево-судинних захворювань. NRF2 також відіграє важливу роль у балансуванні заліза та кальцію, які можуть захистити організм людини від підвищеного рівня заліза. Наприклад, сиртуїн 2 або SIRT2 може регулювати гомеостаз заліза в клітинах шляхом активації NRF2, який, як вважають, необхідний для здорового рівня заліза. NRF2 також може допомогти при серповидно-клітинній хворобі або SCD. Дисфункція NRF2 може бути причиною ендотоксемії, наприклад, при дисбактеріозі або гіпертензії, індукованої лектинами. Nrf2 також може захищати організм людини від пошкодження судинної системи, викликаного амфетаміном.
Бореться з нейрозапаленням
NRF2 може захистити від запалення мозку, яке зазвичай називають нейрозапаленням, і допомогти при цьому. Крім того, NRF2 може допомогти при ряді розладів центральної нервової системи або ЦНС, включаючи:
Хвороба Альцгеймера (AD) – зменшує бета-напругу амілоїду на мітохондрії
Бічний аміотрофічний склероз (БАС)
Хвороба Гентінгтона (HD)
Розсіяний склероз (MS)
Регенерація нервів
Хвороба Паркінсона (ХП) – захищає дофамін
Травма спинного мозку (SCI)
Інсульт (ішемічний та геморагічний) – сприяє гіпоксії
Травма мозку
NRF2 виявив зменшення нейрозапалення у підлітків з розладами аутичного спектру або РАС. Ідебенон правильно поєднується з активаторами NRF2, на відміну від нейрозапалення. NRF2 також може покращити гематоенцефалічний бар’єр, або BBB. Наприклад, активація NRF2 карнозовою кислотою, отриманою з розмарину та шавлії, може перетинати ГЕБ і викликати нейрогенез. Також було продемонстровано, що NRF2 підвищує нейротрофічний фактор мозку, або BDNF.
NRF2 також модулює здатність деяких харчових добавок викликати фактор росту нервів, або NGF, оскільки він також може допомогти з проблемами туману мозку та глутаматом, модулюючи N-метил-D-аспартат, або NMDA-рецептори. Він також може знизити окислювальний стрес від хінолінової кислоти, званої QUIN. Активація NRF2 може захистити від судом, а великі дози можуть зменшити межу нападу. При регулярних дозах стимуляції NRF2 може підвищити когнітивні здібності після судом, знижуючи рівень позаклітинного глутамату в мозку і завдяки його здатності витягувати цистеїн з глутамату і глутатіону.
Знижує депресію
При депресії цілком нормально помічати запалення в мозку, особливо з префронтальної кори та гіпокампу, а також зниження BDNF. У деяких версіях депресії NRF2 може покращувати симптоми депресії, зменшуючи запалення в мозку та підвищуючи рівень BDNF. Здатність агматину зменшувати депресію шляхом підвищення норадреналіну, дофаміну, серотоніну та BDNF в гіпокампі залежить від активації NRF2.
Містить протиракові властивості
NRF2 в рівній мірі є супресором пухлини, як і стимулятором пухлини, якщо його не керувати відповідним чином. NRF2 може захистити від раку, спричиненого вільними радикалами та окислювальним стресом, однак надекспресія NRF2 також може бути виявлена в ракових клітинах. Інтенсивна активація NRF2 може допомогти з різними видами раку. Наприклад, добавка Protandim може зменшити рак шкіри шляхом активації NRF2.
Знімає біль
Хвороба війни в Перській затоці, або GWI, помітна хвороба, яка вражає ветеранів війни в Перській затоці, являє собою сукупність незрозумілих хронічних симптомів, які можуть включати втому, головні болі, біль у суглобах, розлад травлення, безсоння, запаморочення, респіраторні захворювання та проблеми з пам’яттю. NRF2 може покращити симптоми GWI, зменшуючи гіпокампальний та загальне запалення, а також зменшуючи біль. NRF2 може додатково полегшити біль від травми тілесного нерва та покращити пошкодження нервів від діабетичної нейропатії.
Покращує діабет
Високий рівень глюкози, найкраще названий гіперглікемією, викликає окисне пошкодження клітин через порушення функції мітохондрій. Активація NRF2 може захистити організм людини від пошкодження клітини гіперглікемією, тим самим запобігаючи загибелі клітини. Активація NRF2 може додатково захистити, відновити та посилити функцію бета-клітин підшлункової залози, одночасно знижуючи резистентність до інсуліну.
Захищає зір і слух
NRF2 може захистити око від діабетичної ретинопатії. Це також може запобігти утворенню катаракти та захистити фоторецептори від смерті від світла. NRF2 додатково захищає вухо або равлику від стресу та втрати слуху.
Може допомогти при ожирінні
NRF2 може допомогти в боротьбі з ожирінням насамперед завдяки своїй здатності регулювати змінні, які впливають на накопичення жиру в людському тілі. Активація NRF2 за допомогою сульфорафану може призвести до інгібування синтезу жирних кислот, або FAS, і роз’єднаних білків, або UCP, що призводить до меншого накопичення жиру та збільшення кількості коричневого жиру, що характеризується як жир, який включає більше мітохондрій.
Захищає кишечник
NRF2 допомагає захистити кишечник, захищаючи гомеостаз мікробіома кишечника. Наприклад, пробіотики лактобактерій запускають NRF2, щоб захистити кишечник від окисного стресу. NRF2 також може допомогти запобігти виразковий коліт або UC.
Захищає статеві органи
NRF2 може захистити яєчка і захистити кількість сперматозоїдів від шкоди у людей з діабетом. Це також може допомогти при еректильній дисфункції або ЕД. Деякі добавки, що підвищують лібідо, такі як Мукуна, Трибулус і Ашваганда, можуть підвищити сексуальну функцію через активацію NRF2. Інші фактори, які підвищують NRF2, такі як сонячне світло або паростки брокколі, також можуть допомогти покращити лібідо.
Регулює роботу кісток і м’язів
Окислювальний стрес може призвести до зниження щільності і міцності кісток, що є нормальним при остеопорозі. Активація NRF2 може мати здатність покращувати антиоксиданти в кістках і захищати від старіння кісток. NRF2 також може запобігти втраті м’язової маси та посилити м’язову дистрофію Дюшенна, або МДД.
Містить противірусні властивості
Останнє, але не менш важливе, активація NRF2 може в кінцевому підсумку допомогти захистити організм людини від кількох вірусів. У пацієнтів з вірусом денге симптоми були не такими інтенсивними в осіб, які мали більший рівень NRF2, порівняно з особами, які мали менший ступінь NRF2. NRF2 також може допомогти людям, які мають вірус імунодефіциту людини-1 або ВІЛ. NRF2 може захистити від окислювального стресу, викликаного адено-асоційованим вірусом, або AAV, і H. Pylori. Нарешті, корінь Ліндери може пригнічувати вірус гепатиту С за допомогою активації NRF2.
Nrf2, або фактор 2, пов’язаний з NF-E2, є фактором транскрипції, знайденим у людей, який регулює експресію специфічного набору антиоксидантних і детоксикаційних генів. Цей сигнальний шлях активується через окислювальний стрес, оскільки він посилює численні антиоксиданти та ферменти детоксикації печінки фази II для відновлення гомеостазу в організмі людини. Люди пристосовані функціонувати протягом усього стану гомеостазу або рівноваги. Коли організм стикається з окислювальним стресом, Nrf2 активується, щоб регулювати окислення та контролювати стрес, який він викликає. Nrf2 необхідний для запобігання проблем зі здоров'ям, пов'язаних з окислювальним стресом. Д-р Алекс Хіменес, округ Колумбія, CCST Insight
Сульфорафан та його вплив на рак, смертність, старіння, мозок і поведінку, хвороби серця тощо
Ізотіоціанати є одними з найважливіших рослинних сполук, які ви можете отримати у своєму раціоні. У цьому відео я роблю для них найповнішу справу, яку коли-небудь робили. Короткий період уваги? Перейдіть до улюбленої теми, натиснувши один із моментів часу нижче. Повна хронологія нижче.
Ключові розділи:
00:01:14 – Рак і смертність
00:19:04 – Старіння
00:26:30 – Мозок і поведінка
00:38:06 – Підсумок
00:40:27 – Доза
Повний графік:
00:00:34 – Представлення сульфорафану, головна тема відео.
00:01:14 – Споживання овочів хрестоцвітних і зниження смертності від усіх причин.
00:02:12 – Ризик раку передміхурової залози.
00:02:23 – Ризик раку сечового міхура.
00:02:34 – Ризик раку легенів у курців.
00:02:48 – Ризик раку молочної залози.
00:03:13 – Гіпотетична: що робити, якщо у вас уже рак? (інтервенційний)
00:03:35 – Імовірний механізм, який керує асоціативними даними раку та смертності.
00:04:38 – Сульфорафан і рак.
00:05:32 – Докази на тваринах, що показують сильний вплив екстракту паростків брокколі на розвиток пухлин сечового міхура у щурів.
00:06:06 – Вплив прямого прийому сульфорафану у пацієнтів з раком передміхурової залози.
00:07:09 – Біоакумуляція метаболітів ізотіоціаната у фактичній тканині молочної залози.
00:08:32 – Пригнічення стовбурових клітин раку молочної залози.
00:08:53 – Урок історії: ще в Стародавньому Римі стверджували, що капустяні гриби мають оздоровчі властивості.
00:09:16 – Здатність сульфорафану посилювати виведення канцерогену (бензолу, акролеїну).
00:09:51 – NRF2 як генетичний перемикач через елементи антиоксидантної реакції.
00:10:10 – Як активація NRF2 посилює виведення канцерогену через глутатіон-S-кон'югати.
00:10:34 – Брюссельська капуста підвищує глутатіон-S-трансферазу і зменшує пошкодження ДНК.
00:11:20 – Напій з проростків брокколі збільшує виведення бензолу на 61%.
00:15:45 – Споживання хрестоцвітних овочів і смертність від серцево-судинних захворювань.
00:16:55 – порошок паростків брокколі покращує рівень ліпідів у крові та загальний ризик серцевих захворювань у діабетиків 2 типу.
00:19:04 – Початок секції старіння.
00:19:21 – Дієта, збагачена сульфорафаном, збільшує тривалість життя жуків від 15 до 30% (за певних умов).
00:20:34 – Важливість слабкого запалення для довголіття.
00:22:05 – Овочі хрестоцвітних і порошок паростків брокколі, здається, зменшують широкий спектр запальних маркерів у людей.
00:23:40 – Підсумок у середині відео: розділи про рак, старіння
00:24:14 – Дослідження на мишах показують, що сульфорафан може покращити адаптивну імунну функцію в літньому віці.
00:25:18 – Сульфорафан покращив ріст волосся у мишачої моделі облисіння. Зображення на 00:26:10.
00:26:30 – Початок розділу «Мозок і поведінка».
00:27:18 – Вплив екстракту паростків брокколі на аутизм.
00:27:48 – Вплив глюкорафаніну на шизофренію.
00:28:17 – Початок обговорення депресії (правдоподібний механізм та дослідження).
00:31:21 – Дослідження на мишах з використанням 10 різних моделей депресії, викликаної стресом, показало, що сульфорафан так само ефективний, як і флуоксетин (прозак).
00:32:00 – Дослідження показує, що пряме вживання глюкорафаніну мишами так само ефективне для запобігання депресії через модель стресу соціальної поразки.
00:33:01 – Початок відділу нейродегенерації.
00:33:30 – Сульфорафан і хвороба Альцгеймера.
00:33:44 – Сульфорафан і хвороба Паркінсона.
00:33:51 – Сульфорафан і хвороба Хантінгтона.
00:34:13 – Сульфорафан збільшує кількість білків теплового шоку.
00:34:43 – Початок секції черепно-мозкової травми.
00:35:01 – Сульфорафан, введений відразу після ЧМТ, покращує пам’ять (дослідження на мишах).
00:35:55 – Сульфорафан і нейрональна пластичність.
00:36:32 – Сульфорафан покращує навчання на моделі діабету ІІ типу у мишей.
00:37:19 – Сульфорафанова і м’язова дистрофія Дюшенна.
00:37:44 – Інгібування міостатину в клітинах-супутниках м’язів (in vitro).
00:38:06 – Пізнє відео: смертність і рак, пошкодження ДНК, окислювальний стрес і запалення, виділення бензолу, серцево-судинні захворювання, діабет ІІ типу, вплив на мозок (депресія, аутизм, шизофренія, нейродегенерація), шлях NRF2.
00:40:27 – Думки щодо визначення дози паростків брокколі або сульфорафану.
00:41:01 – Анекдоти про проростання в домашніх умовах.
00:43:14 – Про температуру приготування та активність сульфорафану.
00:43:45 – Перетворення сульфорафану з глюкорафаніну кишковими бактеріями.
00:44:24 – Добавки працюють краще в поєднанні з активною мирозиназою з овочів.
00:44:56 – Техніка приготування та овочі хрестоцвітних.
00:46:06 – Ізотіоціанати як зоб.
Коли людський організм стикається з шкідливими внутрішніми та зовнішніми факторами, такими як токсини, клітини повинні швидко активувати свої антиоксидантні здібності, щоб протидіяти окислювальному стресу. Оскільки було встановлено, що підвищений рівень окисного стресу викликає різноманітні проблеми зі здоров’ям, важливо використовувати активацію Nrf2, щоб скористатися її перевагами. Обсяг нашої інформації обмежено хіропрактикою та проблемами здоров’я хребта. Щоб обговорити тему, зверніться до доктора Хіменеса або зв’яжіться з нами за адресою�915-850-0900.
Куратор доктор Алекс Хіменес
Додаткова тема для обговорення: «Гострий біль у спині».
Біль у спині� є однією з найпоширеніших причин інвалідності та пропущених робочих днів у всьому світі. Біль у спині є другою за поширеністю причиною відвідувань лікаря, переважаючи лише інфекції верхніх дихальних шляхів. Приблизно 80 відсотків населення відчувають біль у спині хоча б раз у житті. Хребет – це складна структура, що складається з кісток, суглобів, зв’язок і м’язів, серед інших м’яких тканин. Через це травми та/або загострення стану, такі як�грижі диски, може зрештою призвести до симптомів болю в спині. Спортивні травми або травми в автомобільній катастрофі часто є найчастішою причиною болю в спині, однак іноді найпростіші рухи можуть мати хворобливі наслідки. На щастя, альтернативні варіанти лікування, такі як хіропрактика, можуть допомогти полегшити біль у спині за допомогою корекції хребта та ручних маніпуляцій, що в кінцевому підсумку покращує полегшення болю. �
Сульфорафан це фітохімічна речовина, речовина в групі ізотіоціанатних сіркоорганічних сполук, що міститься в хрестоцвітних овочах, таких як брокколі, капуста, цвітна та брюссельська капуста. Його також можна знайти в бок-чої, капусті, капусті, зелені гірчиці та крес-салата. Дослідження показали, що сульфорафан може допомогти запобігти різним типам раку активація виробництва Nrf2, або ядерний фактор, пов’язаний з еритроїдом 2, фактор транскрипції, який регулює захисні антиоксидантні механізми, які контролюють реакцію клітини на окислювачі. Мета наступної статті — описати функцію сульфорафану.
абстрактний
Антиоксидантна система KEAP1-Nrf2-ARE є основним засобом, за допомогою якого клітини реагують на окислювальний і ксенобіотичний стрес. Сульфорафан (SFN), електрофільний ізотіоціанат, отриманий з овочів хрестоцвітих, активує шлях KEAP1-Nrf2-ARE і став молекулою, що представляє інтерес у лікуванні захворювань, у яких головну етіологічну роль відіграє хронічний окислювальний стрес. Тут ми демонструємо, що мітохондрії культивованих клітин пігментного епітелію сітківки сітківки (RPE-1) людини, оброблених SFN, піддаються гіперфузії, яка не залежить як від Nrf2, так і від його цитоплазматичного інгібітора KEAP1. Повідомляється, що злиття мітохондрій є цитопротекторним шляхом інгібування утворення пор в мітохондріях під час апоптозу, і відповідно до цього ми показуємо Nrf2-незалежний цитозахист клітин, оброблених SFN, підданих впливу індуктора апоптозу, стауроспорину. Механічно SFN пом’якшує залучення та/або утримання розчинного фактора поділу Drp1 в мітохондріях і пероксисомах, але не впливає на загальну кількість Drp1. Ці дані демонструють, що корисні властивості SFN виходять за межі активації системи KEAP1-Nrf2-ARE і вимагають подальшого допиту, враховуючи поточне використання цього агента в численних клінічних дослідженнях.
Сульфорафан - це Nrf2-незалежний інгібітор мітохондріального поділу
Сульфорафан (SFN) — ізотіоціанатна сполука, що одержується в раціоні, найчастіше з овочів хрестоцвітих [56]. Він утворюється в рослинах як ксенобіотична відповідь на хижацтво через везикулярне вивільнення гідролітичного ферменту мірозинази з пошкоджених клітин; цей фермент перетворює глюкозинолати в ізотіоціанти [42]. Протягом останніх двох десятиліть SFN широко охарактеризований через його протиракові, антиоксидантні та антимікробні властивості [57]. Значна частина цієї ефективності пояснюється здатністю SFN модулювати сигнальний шлях KEAP1-Nrf2-антиоксидантної відповіді (ARE), хоча були виявлені додаткові дії сполуки, включаючи інгібування активності гістондеацетилази та прогресування клітинного циклу [ 29]. Nrf2 є головним антиоксидантним фактором транскрипції і в умовах гомеостазу його стабільність пригнічується під дією цитоплазматичного комплексу убіквітинлігази Cullin3KEAP1 [20]. Зокрема, Nrf2 залучається до лігази Cullin3KEAP1 шляхом зв’язування з димерним субстратним адаптером KEAP1 і згодом модифікується ланцюгами polyUb, які спрямовані на фактор транскрипції для опосередкованої протеасомою деградації. Цей конститутивний оборот обмежує період напіввиведення Nrf2 в клітинах без стресу до ~15 хв [30], [33], [46], [55]. У відповідь на численні типи стресу, зокрема окислювальний стрес, KEAP1, багатий цистеїном білок, діє як окислювально-відновний датчик, а окислювальна модифікація критичних цистеїнів, зокрема C151, KEAP1 відокремлює Nrf2-KEAP1 від CUL3, таким чином запобігаючи деградації Nrf2 [ 8], [20], [55]. Примітно, що SFN і, можливо, інші активатори Nrf2 імітують окислювальний стрес шляхом модифікації C151 KEAP1, наприклад [21]. Стабілізація Nrf2 забезпечує його транслокацію в ядро, де він індукує експресію батареї антиоксидантних і детоксикаційних генів фази II. Nrf2 зв’язується з елементами промотору антиоксидантної відповіді (ARE) своїх споріднених цільових генів шляхом гетеродимеризації невеликими білками Maf [19]. Ця система представляє динамічну та чутливу реакцію на непрямі антиоксиданти, такі як SFN, вільні радикали, що утворюються мітохондріями [16], або інші фізіологічні джерела окисного стресу [41].
Мітохондрії – це динамічні субклітинні органели, які регулюють безліч клітинних функцій, починаючи від виробництва АТФ і внутрішньоклітинної буферизації кальцію до окислювально-відновної регуляції та апоптозу [13], [49]. Мітохондрії також є основним джерелом активних форм кисню (АФК) всередині клітини. Тому правильна регуляція мітохондріальної функції необхідна для оптимізації виробництва АТФ для задоволення потреб клітин, одночасно зводячи до мінімуму потенційно шкідливі ефекти надмірного виробництва вільних радикалів. Критичною вимогою для тонкої модуляції мітохондріальної функції є здатність мітохондрій функціонувати як незалежно як біохімічні машини, так і як частина величезної мережі, що реагує.
Морфологія і функція мітохондріальної мережі визначаються регульованим балансом між поділом і злиттям. Ділення мітохондрій необхідне для успадкування мітохондрій дочірньою клітиною під час поділу клітини [28], а також для селективної аутофагічної деградації деполяризованих або пошкоджених мітохондрій, що називається мітофагією [1]. І навпаки, злиття необхідне для комплементації мітохондріальних геномів і спільного використання компонентів ланцюга транспорту електронів між сусідніми мітохондріями [54]. На молекулярному рівні поділ і злиття мітохондрій регулюються великими динаміноподібними ГТФазами. Три ферменти головним чином регулюють злиття: мітофузини 1 і 2 (Mfn1/2) є двопрохідними зовнішніми мембранними білками, які опосередковують злиття зовнішньої мембрани за допомогою гетеротипних взаємодій між сусідніми мітохондріями [15], [25], [37], тоді як OPA1 є внутрішнім мембранний білок, який одночасно забезпечує зв’язність матриці, регулюючи злиття внутрішніх мембран [5]. Активність ГТФази всіх трьох білків необхідна для надійного злиття [5], [18], а OPA1 додатково регулюється комплексним протеолізом у внутрішній мембрані мітохондрій протеазами OMA1 [14], PARL [6] і YME1L [45]. ]. Важливо, що потенціал інтактної мітохондріальної мембрани необхідний для ефективного злиття, щоб придушити інтеграцію пошкоджених і здорових мітохондрій [26].
Ділення мітохондрій в основному каталізується цитозольним білком, який називається білком 1, пов’язаним з Dynamin (Drp1/DNM1L). Drp1 рекрутується з цитозолю до передбачуваних місць поділу на зовнішній мембрані мітохондрій [43]. Основними рецепторами для Drp1 на зовнішній мембрані є мітохондріальний фактор поділу (Mff) [32] і, меншою мірою, Fission 1 (Fis1) [51]. Крім того, було виявлено рецептор-приманку, MIEF1/MiD51, який діє для подальшого обмеження активності білка Drp1 у потенційних місцях поділу [58]. Після прикріплення до зовнішньої мембрани мітохондрії Drp1 олігомеризується в спіралеподібні структури навколо тіла мітохондрії, а потім використовує енергію, отриману від гідролізу GTP, для опосередкування фізичного розщеплення зовнішньої та внутрішньої мембран мітохондрій [17]. Трубочки, отримані з ендоплазматичної мережі, діють як початковий констриктор мітохондрій до олігомеризації Drp1, підкреслюючи відкриття, що незвужені мітохондрії ширші, ніж дозволена окружність завершеної спіралі Drp1 [12]. Динаміка актину також важлива для взаємодії ЕР-мітохондрій, які передують поділу мітохондрій [24]. На додаток до своєї ролі в поділі мітохондрій, Drp1 каталізує поділ пероксисом [40].
Drp1 дуже схожий на добре охарактеризований білок динамін тим, що обидва білки містять N-кінцевий домен GTPase, середній домен, який є критичним для самоолігомеризації, і C-кінцевий ефекторний домен GTPase [31]. Drp1 досягає селективності для мітохондріальних мембран завдяки комбінації взаємодій з його рецепторними білками Mff і Fis1, а також завдяки спорідненості до мітохондрій-специфічного фосфоліпідного кардіоліпіну через унікальний В-вставний домен Drp1 [2]. Drp1 зазвичай існує як гомотетрамер у цитоплазмі, а збірка вищого порядку в місцях поділу мітохондрій опосередковується середнім доменом Drp1 [3].
Враховуючи неявний зв’язок між мітохондріальною функцією та шляхом KEAP1-Nrf2-ARE, ми досліджували вплив активації Nrf2 на структуру та функцію мітохондрій. Тут ми демонструємо, що SFN індукує гіперфузію мітохондрій, яка несподівано не залежить як від Nrf2, так і від KEAP1. Цей ефект SFN відбувається через пригнічення функції Drp1. Далі ми демонструємо, що SFN надає стійкість до апоптозу, яка не залежить від Nrf2 і імітує те, що спостерігається в клітинах, збіднених Drp1. Ці дані разом вказують на те, що на додаток до стабілізації та активації Nrf2, SFN модулює мітохондріальну динаміку та зберігає придатність і виживання клітин.
результати
Сульфорафан викликає Nrf2/KEAP1-незалежну гіперфузію мітохондрій
У ході вивчення впливу активації Nrf2 на динаміку мітохондріальної мережі ми виявили, що обробка імморталізованих клітин пігментного епітелію сітківки людини (RPE-1) сульфорафаном (SFN), потужним активатором передачі сигналів Nrf2, індукує міцне злиття мітохондріальну мережу в порівнянні з контрольними клітинами, обробленими транспортним засобом (рис. 1A і B). Морфологія мітохондрій у цих клітинах дуже нагадувала морфологію мітохондрій у клітинах, збіднених siRNA ендогенного Drp1, основного фактора мітохондріального поділу (рис. 1A). Цей результат викликав інтригуючу ідею, що стан поділу та злиття мітохондрій безпосередньо реагує на рівні Nrf2 в клітині. Однак стимуляція клітин іншими стабілізаторами та активаторами Nrf2, такими як інгібітор протеасоми MG132, прооксидант tBHQ, або нокдаун інгібітора Nrf2 KEAP1 не викликали злиття мітохондрій (рис. 1A і B). Стабілізація Nrf2 за допомогою цих маніпуляцій була підтверджена вестерн-блоттингом для ендогенного Nrf2 (рис. 1C). Крім того, експресія Nrf2 була необхідна для SFN-індукованого мітохондріального злиття, оскільки нокдаун ендогенного Nrf2 за допомогою siRNA не зміг протистояти цьому фенотипу (рис. 1D�F). Оскільки SFN стимулює шлях KEAP1-Nrf2-ARE шляхом ковалентної модифікації залишків цистеїну KEAP1 [21], ми знищили KEAP1, щоб вирішити, чи стимулюється SFN-індукована гіперфузія мітохондрій через KEAP1-залежний, але незалежний шлях від Nrf2. Однак виснаження KEAP1 також не вдалося скасувати індукований SFN мітохондріальний злиття (рис. 1G�I). Фактично, SFN змінив морфологію професійного поділу, викликану виснаженням KEAP1 (рис. 1G, панель b проти панелі d). Ці результати вказують на те, що лікування SFN викликає злиття мітохондрій незалежно від канонічного шляху KEAP1-Nrf2-ARE, і підштовхнуло нас до питання, чи впливає SFN безпосередньо на компоненти мітохондріального поділу або механізму злиття.
Малюнок 1 SFN індукує Nrf2/KEAP1-незалежне мітохондріальне злиття. (A) Клітини RPE-1 трансфікували вказаними siRNA і через 3 дні обробляли ДМСО або активаторами Nrf2 SFN (50 мкМ), MG132 (10 мкМ) або tBHQ (100 мкМ) протягом 4 год. Мітохондрії (червоні) мічені антитілом до Tom20, а ядра (сині) забарвлені DAPI. (B) Графік, що показує кількісне визначення морфології мітохондрій з (A). >50 клітин на стан оцінювали наосліп. (C) Репрезентативні вестерн-блоти з (A). (D) Клітини RPE-1 трансфікували 10 нМ siRNA і через 3 дні обробляли SFN протягом 4 годин перед фіксацією та фарбуванням, як у (A). (E) Графік, що показує кількісне визначення мітохондріального фенотипу з (D). >100 клітин на стан оцінювали сліпим способом. (F) Репрезентативні вестерн-блоти з (D). (G) Клітини трансфікували та обробляли, як у (D), siCON або siKEAP1. (H) Клітини з (G) оцінювали як у (B) і (E) на основі мітохондріальної морфології. (I) Репрезентативні вестерн-блоти з (G). Дані в (B), (E) і (H) були зібрані з 3 незалежних експериментів, кожен і статистичну значущість визначали двостороннім t-критерієм Стьюдента. Смужки помилок відображають +/- SD (Для інтерпретації посилань на колір у легенді цього малюнка читач звертається до веб-версії цієї статті).
Виходячи з висновку, що лікування SFN викликає гіперфузію мітохондрій, ми прийшли до висновку, що цей фенотип був або наслідком надмірної активності злиття, або пригніченням активності поділу. Щоб розрізнити ці дві можливості, ми порівняли морфологію пероксисом у присутності та відсутності SFN. Пероксисоми подібні до мітохондрій тим, що вони є динамічними органелами, форма і довжина яких постійно змінюються [44]. Пероксисоми містять як Fis1, так і Mff у своїй зовнішній мембрані і, як наслідок, є мішенями для Drp1-опосередкованого поділу [22], [23]. Однак пероксисоми не використовують механізм злиття мітохондріальної мережі і, отже, не зазнають злиття [39]. Швидше, пероксисомальному поділу протистоїть подовження існуючих пероксисом за рахунок de novo додавання мембран і білків [44]. Оскільки в пероксисомах відсутні Mfn1/2 та OPA1, ми прийшли до висновку, що якщо SFN активує механізм злиття, а не інгібує механізм поділу, довжина пероксисом не вплине. У клітинах, оброблених носієм, пероксисоми зберігаються у вигляді коротких круглих точковидних органел (рис. 2, панелі b і d). Однак лікування SFN збільшило довжину пероксисом приблизно в 2 рази порівняно з контрольними клітинами (рис. 2, панелі f і h). Крім того, багато пероксисом були защемлені поблизу центру, що вказує на потенційний дефект розрізу (рис. 2, панель h, наконечники стрілок). Аналогічно, пероксисоми в клітинах, трансфікованих миРНК Drp1, були аномально довгими (рис. 2, панелі j і l), що підтверджує, що Drp1 необхідний для пероксисомального поділу, і припускає, що лікування SFN викликає мітохондріальні та пероксисомні фенотипи, порушуючи механізм поділу.
Малюнок 2 SFN викликає подовження пероксисом. (A) Клітини RPE-1 трансфікували 10 нМ зазначеної siRNA і через 3 дні обробляли ДМСО або 50 мкМ SFN протягом 4 годин. Пероксисоми (зелені) були мічені антитілом проти PMP70, мітохондрії за допомогою MitoTracker (червоний), а ДНК зафарбовували DAPI. Праворуч показані збільшені вставки пероксисом (панелі d, h і l), щоб полегшити візуалізацію змін морфології, спричинених виснаженням SFN та Drp1. Стрілки виділяють точки звуження. (Для інтерпретації посилань на колір у легенді цього малюнка читач звертається до веб-версії цієї статті).
Далі ми визначили, як SFN обмежує функцію Drp1. Можливості включали зниження рівня експресії, рекрутування/утримання в мітохондріях, олігомеризацію або ферментативну активність GTPase. Дефіцит будь-якого з них призведе до зниження мітохондріального поділу та гіперфузії. Ми не виявили відтворюваних змін у рівнях білка Drp1 після лікування SFN (рис. 1C і 3A), і тому дійшли висновку, що SFN не змінює стабільність або експресію Drp1, відповідно до того, що Drp1 має період напіввиведення >10 годин [50] і наші лікування SFN мають меншу тривалість. Далі ми дослідили, чи впливає SFN на залучення або утримання Drp1 в мітохондріях. Дослідження фракціонування показали, що SFN викликав втрату Drp1 з мітохондріальної фракції (рис. 3A, доріжки 7�8 і рис. 3B). Як повідомлялося раніше [43], лише незначна частина Drp1 (~3%) пов’язана з мітохондріальною мережею в будь-який момент часу під час стаціонарних умов, при цьому більшість ферменту знаходиться в цитоплазмі (рис. 3A, доріжки 5�8 ). Ці дані фракціонування були підтверджені за допомогою аналізу спільної локалізації, який показав зменшення на 40% у мітохондріях точкових вогнищ Drp1 після обробки SFN (рис. 3C і D). Разом ці дані вказують на те, що злиття мітохондрій, викликане SFN, принаймні частково зумовлене ослабленою асоціацією Drp1 з мітохондріями. Наші дані не розрізняють, чи впливає SFN на рекрутацію мітохондрій від утримання Drp1 у мітохондріях, чи обидва, оскільки аналіз ендогенного Drp1 не піддавався візуалізації GTPази за допомогою мікроскопії живих клітин.
Малюнок 3 SFN викликає втрату Drp1 з мітохондрій. (A) Субклітинне фракціонування клітин RPE-1 після 4 год DMSO або SFN. Цільноклітинні лізати (WCL), ядерні (Nuc), цитозольні (Cyto) та неочищені мітохондріальні (Mito) фракції розділяли за допомогою SDS-PAGE та обробляли для вестерн-блоттингу із зазначеними антитілами. Міграція маркерів молекулярної маси вказана зліва. (B) Графіки, що показують денситометричне кількісне визначення Drp1 у зазначених фракціях з (A). (C) Клітини RPE-1 трансфікували 10 нМ siCON або siDrp1 і через 3 дні обробляли ДМСО або SFN протягом 4 годин. Drp1 (зелений) візуалізували за допомогою антитіла до Drp1, мітохондрії — за допомогою MitoTracker (червоний), а ядра — за допомогою DAPI (синій). (D) Автоматичний аналіз спільної локалізації сигналу Drp1 і MitoTracker з (C). Дані в (B) і (D) були зібрані з 3 і 5 незалежних експериментів відповідно, а статистичну значущість визначали двостороннім t-критерієм Стьюдента. Стрічки помилок відображають +/- SD, а зірочки позначають статистичну значущість. (Для інтерпретації посилань на колір у легенді цього малюнка читач звертається до веб-версії цієї статті).
Сульфорафан забезпечує захист від апоптозу, індукованого стауроспортином, незалежно від Nrf2
Попередня робота показала, що поділ мітохондрій є дозволеним у формуванні пір у зовнішній мітохондріальній мембрані, утворених Bax/Bak під час апоптозу [11]. Показано, що Drp1 вибірково залучається до мітохондрій під час апоптозу [11], і, відповідно до цього, фрагментовані мітохондрії спостерігалися на початку процесу [27]. І навпаки, вважають, що інгібування поділу мітохондрій пригнічує апоптоз, блокуючи утворення пор зовнішньої мембрани, які забезпечують вивільнення цитохрому c [53]. Відповідно, стимуляція злиття мітохондрій затримує прогресування апоптозу, індукованого сполуками, включаючи стауроспорин (STS) [14]. Щоб визначити, чи захищає SFN клітини RPE-1 від апоптозу, опосередкованого STS, і якщо так, то чи потрібен для цього Nrf2, ми створили аналіз, щоб легко індукувати розщеплення поліАДФ рибозополімерази (PARP), субстрату активованої каспази-3 і остаточного маркера апоптоз. Обробка клітин RPE-1 1 мкМ STS протягом 6 годин викликала лише дуже помірне розщеплення PARP, але цьому було попереджено спільне лікування SFN (наприклад, рис. 4A, доріжка 3 проти 4). Щоб підвищити надійність цього аналізу, ми додатково сенсибілізували клітини до апоптозу, індукованого STS, попередньо обробивши їх siRNA, спрямованою на антиапоптотичний фактор, Bcl-XL. Ця попередня обробка зменшувала експресію Bcl-XL і помітно сприяла розщепленню PARP як функції часу впливу STS (рис. 4B, порівняйте доріжку 2 з доріжками 4÷10). Важливо, що 2 години попередньої обробки SFN пом’якшили розщеплення PARP в клітинах, підданих STS (рис. 4C, доріжка 3 проти 4 і доріжка 5 проти 6). Аналогічно, клітини, стабільно виснажені Nrf2 за допомогою CRISPR/Cas9, були порівняно захищені від токсичності STS попередньою обробкою SFN (рис. 4C, доріжка 11 проти 12 і смуга 13 проти 14 і рис. 4D). Цей захист спостерігали, використовуючи як розщеплення PARP (рис. 4C і D), так і клітинну морфологію (рис. 4E) як показання. Ефективність виснаження Nrf2 за допомогою CRISPR/Cas9 була підтверджена вестерн-блоттингом (рис. 4C, Nrf2 блот). Як було передбачено, виснаження клітин Drp1, яке також дає фенотип гіперфузії (рис. 1A), також блокує розщеплення PARP у відповідь на STS порівняно з контрольними клітинами, інкубованими з SFN (рис. 4F та G). Разом ці висновки узгоджуються з тим, що SFN забезпечує захист від апоптозу через його здатність обмежувати функцію Drp1, незалежно від стабілізації та активації Nrf2.
Рисунок 4. Цитопротекторні ефекти SFN не залежать від експресії Nrf2 (A). Клітини RPE-1 попередньо обробляли ДМСО або 50 мкМ SFN протягом 2 год перед обробкою ДМСО, 1 мкМ стауроспорином (STS) або 50 ? М етопозид протягом 6 год і обробляли для вестерн-блоттингу проти PARP. (B) Клітини RPE-1 трансфікували 2.5 нМ siCON, 1 нМ siBcl-XL або 2.5 нМ siBcl-XL і через 3 дні обробляли ДМСО або 1 мкМ STS протягом 2, 4 або 6 годин. Показані репрезентативні вестерн-блоти, а зліва вказана міграція маркерів молекулярної маси. (C) Клітини RPE-9 дикого типу (Nrf2WT), генеровані CRISPR/Cas2 (Nrf2WT) і нокаутовані Nrf1 (Nrf1KO), трансфікували 3 нМ siBcl-XL і через 50 дні попередньо обробляли ДМСО або 2 мкМ SFN протягом 1 год. . Згодом клітини обробляли 2 мкМ STS протягом 4, 6 або 3 годин. Показані репрезентативний вестерн-блоттинг із зазначеними антитілами. (D) Кількісна оцінка розщепленого PARP у відсотках від загального PARP (розщеплений+нерозщеплений) з 2 незалежних експериментів. Важливо, що рівні розщепленого PARP були порівнянними, незалежно від того, експресують клітини Nrf20 чи ні, що вказує на те, що захист SFN від STS не залежить від фактора транскрипції. (E) 65X фазово-контрастні зображення, зроблені безпосередньо перед збиранням лізатів з (C). Масштабна смуга = 1 м. (F) Репрезентативні вестерн-блоти, які демонструють, що виснаження Drp1 забезпечує майже порівнянний захист від STS як лікування SFN. Клітини RPE-1 трансфікували 10 нМ siBcl-XL і додатково трансфікували 10 нМ siCON або 1 нМ siDrp3. Через 4 дні клітини siCON попередньо обробляли SFN, як у (A) і (C), а потім піддавали STS протягом 3 годин перед збиранням та обробкою для вестерн-блоттингу із зазначеними антитілами. (G) Те саме, що (D) для даних, представлених у (F), зібраних з XNUMX незалежних експериментів. Смужки помилок відображають +/- SEM
Обговорення
Ми виявили, що SFN модулює динаміку мітохондріального поділу/злиття незалежно від його впливу на шлях KEAP1-Nrf2-ARE. Це інтригує через передбачуваний зв’язок між мітохондріальною дисфункцією та продукцією АФК та необхідністю придушення вільних радикалів, отриманих з мітохондрій, шляхом активації Nrf2. Цей додатковий функціональний вплив SFN має потенційне значення з огляду на більш ніж 30 клінічних випробувань, які в даний час проводяться з тестування SFN для лікування різноманітних захворювань, включаючи рак передміхурової залози, обструктивну хворобу легенів та серповидно-клітинну хворобу [7], [10], [ 47].
Оскільки SFN є ізотіоціанатом [56] і він активує передачу сигналів Nrf2 шляхом безпосереднього ацилювання критичних цистеїнів KEAP1 для придушення деградації Nrf2 [21], випливає, що SFN виявляє свої ефекти про-злиття, модулюючи активність фактора поділу або злиття за допомогою модифікації цистеїну. . Наші дані впевнено підтверджують, що Drp1 негативно регулюється SFN, хоча залишається з’ясувати, чи є GTPase прямою мішенню ацилювання. Незважаючи на цей пробіл у знаннях, функція Drp1 явно порушується SFN, оскільки і мітохондрії, і пероксисоми стають гіперфузією у відповідь на лікування SFN, і ці органели спільно використовують Drp1 для відповідних подій розщеплення [38]. Крім того, SFN зменшує кількість Drp1, який локалізується та накопичується в мітохондріях (рис. 3). Оскільки наші експерименти проводилися з усіма ендогенними білками, наше виявлення Drp1 в місцях поділу мітохондрій відбувається в стаціонарних умовах, і, отже, ми не можемо відрізнити рекрутинг від дефекту утримання ферменту, викликаного SFN. Крім того, ми не можемо виключити можливість того, що SFN ацилює рецептор у мітохондріях (Fis1 або Mff), щоб блокувати рекрутинг Drp1, ми підозрюємо, що Drp1 безпосередньо модифікований. Drp1 містить дев'ять цистеїнів, вісім з яких знаходяться в середньому домені, необхідному для олігомеризації [3], і один з яких знаходиться в ефекторному домені GTPase (GED) на C-кінці Drp1. Пряме ацилювання будь-якого з цих цистеїнів може викликати дефект активності Drp1 і, отже, лежить в основі впливу SFN на мітохондріальну динаміку. Примітно, що попередні роботи припускають, що дефекти олігомеризації та каталітичної активності можуть скасувати утримання Drp1 в мітохондріях [52]. Cys644 в домені GED є особливо привабливою мішенню на основі попередніх робіт, які показують, що мутація цього цистеїну фенокопіює мутації, які погіршують активність Drp1 GTPase [4], і що цей конкретний цистеїн модифікується тіол-реактивними електрофілами [9]. Вирішення цього невирішеного питання вимагатиме мас-спектрометричної перевірки. Таким чином, ми визначили нову цитопротекторну функцію для клінічно релевантної сполуки SFN. На додаток до активації головного антиоксидантного фактора транскрипції Nrf2, SFN сприяє злиттю мітохондрій і пероксисом, і цей ефект не залежить від Nrf2. Механізм, що лежить в основі цього явища, включає зниження функції GTPase Drp1, первинного медіатора мітохондріального та пероксисомного поділу. Основним наслідком SFN-опосередкованого злиття мітохондрій є те, що клітини стають стійкими до токсичних ефектів індуктора апоптозу стауроспорину. Ця додаткова цитопротекторна дія SFN може бути особливо корисною для численних нейродегенеративних захворювань, для яких вік є провідним фактором ризику (наприклад, хвороба Паркінсона, хвороба Альцгеймера, вікова макулярна дегенерація), оскільки ці захворювання були пов’язані з апоптозом і зменшували рівні та/або порушення регуляції Nrf2 [35], [36], [48].
Матеріали та методи
Аналіз апоптозу
Клітини висівали та трансфікували siRNA, як зазначено нижче. Клітини попередньо обробляли 50 мкМ сульфорафаном протягом 2 год, щоб індукувати злиття мітохондрій, а потім обробляли 1 мкМ стауроспорином для індукування апоптозу. Під час збору середовищі збирали в окремі пробірки та піддавали високошвидкісному центрифугування для осадження апоптотичних клітин. Цю клітинну гранулу об'єднували з прилиплими клітинами і розчиняли в 2-кратному концентрованому буфері Леммлі. Зразки піддавали вестерн-блоттингу проти PARP.
Генерація конструкцій CRISPR/Cas9
Щоб створити LentiCRISPR/eCas9 1.1, LentiCRISPR v2 (addgene #52961) спочатку було вирізано з Age1 і BamH1. Далі SpCas9 з eSpCas9 1.1 (addgene #71814) був ампліфікований ПЛР з Age1 і BamH1, використовуючи наступні праймери (прямий AGCGCACCGGTTCTAGAGCGCTGCCACCATGGACTATAAGGACCACGAC, зворотний AAGCGGACCACGAC, зворотний AAGCGGATCCCCTTGCC, розрізаний вищевказаний вектор на gTTGCGGATCCCGCTTCGTTGTTGTTGTTGTTG Послідовності sgRNA були визначені за допомогою Benchling.com. Параметри були встановлені так, щоб націлюватися на послідовність кодування з найвищими результатами на цільовому рівні та найнижчими позацільовими. Наступні послідовності (направл Юща послідовність підкреслені, HS sgNFE2L2 # 1 сенс CACCGCGACGGAAAGAGTATGAGC, антисмислового AAACGCTCATACTCTTTCCGTCGC; нд sgNFE2L2 # 2 сенсу CACCGGTTTCTGACTGGATGTGCT, антисмислової AAACAGCACATCCAGTCAGAAACC; нд sgNFE2L2 # 3 сенсу CACCGGAGTAGTTGGCAGATCCAC, антисмислової AAACGTGGATCTGCCAACTACTCC) відпалювали і лігували в BsmB1 вирізати LentiCRISPR / eCas9 1.1. Клітини RPE-1, інфіковані лентивірусом, відбирали за допомогою пуроміцину та підтримували у вигляді об’єднаної популяції. Нокаут був підтверджений імунофлюоресценцією та вестерн-блоттингом.
Культура клітин і трансфекції
Пігментні епітеліальні клітини сітківки сітківки людини, трансформовані теломеразою (RPE-1) (ATCC), культивували в модифікованому середовищі Дульбекко (DMEM), що містить 1 г/л глюкози, доповненої пеніциліном, стрептоміцином, 1X коктейлем незамінних амінокислот (Life Technologies), і 10% фетальної бичачої сироватки (Life Technologies). Для трансфекції siRNA 30,000 35,000–10 0.3 клітин/мл висівали протягом ночі. Клітини отримували 1 нМ siRNA, розведену в DMEM без сироватки та об’єднану з 2% реагентом для трансфекції інтерферину (PolyPlus). Для сенсибілізації апоптозу клітини отримували 3 нМ миРНК Bcl-XL. Клітини збирали через XNUMX дні після трансфекції.
Хімічні речовини, антитіла та олігомінельні РНК
Антитіла проти ?-тубуліну (Cell Signaling), ?-тубуліну (Sigma), Drp1 (BD Biosciences), KEAP1 (Proteintech), Lamin B1 (Abcam), PARP (Cell Signaling), PMP70 (Abcam) і Tom20 (BD Biosciences ) використовували в розведеннях 1:1000 для вестерн-блоттингу та для імунофлуоресценції. Власні кролячі антитіла анти-Nrf2 використовували о 1:2000 для вестерн-блоттингу [34], [59]. Сульфорафан (Sigma) і стауроспорин (Tocris) використовували при 50 мкМ і 1 мкМ відповідно. siRNA проти Drp1 (Dharmacon), Nrf2 (Dharmacon), KEAP1 (Cell Signaling) і Bcl-XL (Cell Signaling) використовували при 10 нМ, якщо не зазначено інше.
Імунофлуоресценція та маркування in vivo
Клітини, засіяні на покривних скель 18 мм, обробляли носієм або препаратом, фіксували в 3.7% формальдегіді і потім пермеабілізували в 0.2% Triton X-100/PBS на льоду протягом 10 хв. Первинні антитіла інкубували в 3% бичачого сироваткового альбуміну (BSA) у PBS протягом ночі при 4 °C. Після промивання PBS клітини інкубували протягом 1 години у відповідних видів, Alexa488- або Alexa546-, кон'югованих вторинних антитілах (розведених 1:1000) і 0.1 мкг/мл DAPI (Sigma) у 3% BSA/PBS. Мітохондрії візуалізували за допомогою імунофлуоресценції проти Tom20 або шляхом інкубації клітин у 200 нМ MitoTracker Red CMXRos (Molecular Probes, Inc.) у безсироватковій DMEM протягом 30 хвилин при 37 °C перед фіксацією.
Мікроскопія та аналіз зображень
Імунофлуоресцентні зразки розглядали на конфокальному мікроскопі LSM710 (Carl Zeiss). Мікрофотографії були зроблені за допомогою об’єктів з масляним зануренням 63X або 100X, а зображення були скориговані та покращені за допомогою Adobe Photoshop CS6. Аналіз спільної локалізації проводили з використанням функції спільної локалізації Carl Zeiss LSM710 з порогами, встановленими вручну, незважаючи на ідентичність зразків. Масштабні смуги на всьому протязі, якщо не вказано інше, становлять 10 м. Морфологію мітохондрій оцінювали шляхом сліпого оцінювання. Якщо мітохондрії клітини зберігалися як множинні круглі дискримінаційні точки, клітина оцінювалася як «розщеплення». Якщо окремі мітохондрії були нерозрізнені, а вся мітохондріальна мережа здавалася безперервною, клітина оцінювалася як «злиття». Усі інші клітини, включаючи ті з кластерними мітохондріями, були оцінені як «проміжні».
Субклітинні фракціонування
Клітини RPE-1 вирощували до злиття. Після промивання PBS клітини центрифугували при 600°g протягом 10 хв і ресуспендували в 600 мкл ізоляційного буфера (210 мМ манітол, 70 мМ сахароза, 5 мМ MOPS, 1 мМ EDTA pH 7.4+1 мМ PMSF). Суспензію лізували 30 разів у гомогенізаторі Dounce. Фракція гомогенату зберігалася як «лізат цільної клітини». Залишок піддавали центрифугування при 800°g протягом 10 хв до осадження ядер. Супернатанти піддавали центрифугування при 1500°g протягом 10 хв для очищення залишилися ядер і нелізованих клітин. Цей супернатант піддавали центрифугування при 15,000ºg протягом 15 хв для осадження мітохондрій. Супернатант зберігали у вигляді «цитозольної фракції». Осад обережно промивали PBS і ресуспендували в ізоляційному буфері. Концентрацію білка в кожній фракції вимірювали за допомогою аналізу біцинхонинової кислоти (BCA), а еквівалентні кількості білка розділяли за допомогою SDS-PAGE.
Вестерн Блотінг
Клітини промивали в PBS і розчиняли в 2-кратному концентрованому розчинному буфері Леммлі (100 мМ Трис [pH 6.8], 2% SDS, 0.008% бромфенолового синього, 2% 2-меркаптоетанолу, 26.3% гліцерину і 0.001% PYO). Лізати кип’ятили протягом 5 хв перед завантаженням у поліакриламідні гелі додецилсульфату натрію (SDS). Білки переносили на нітроцелюлозні мембрани, і мембрани блокували на 1 годину в 5% молоці/TBST. Первинні антитіла розводили у 5% молоці/TBST та інкубували з блоттингом протягом ночі при 4 °C. Вторинні антитіла, кон’юговані з пероксидазою хрону (HRP), розводили в 5% молоці/TBST. Блоти були оброблені з посиленою хемілюмінесценцією, а денситометричні кількісні оцінки проводили за допомогою програмного забезпечення ImageJ.
Сульфорафан — це хімічна речовина із ізотіоціанатної колекції сіркоорганічних речовин, одержуваних із овочів хрестоцвітних, включаючи брокколі, капусту, цвітну капусту, капусту та капусту, серед інших. Сульфорафан утворюється, коли фермент мирозиназа перетворює глюкорафанін, глюкозинолат, у сульфорафан, також відомий як сульфорафан-глюкозинолат. Паростки брокколі та цвітна капуста мають найвищу концентрацію глюкорафаніну або попередника сульфорафану. Дослідження показали, що сульфорафан підвищує антиоксидантні властивості людського організму для запобігання різних проблем зі здоров’ям. Д-р Алекс Хіменес, округ Колумбія, CCST Insight
Сульфорафан та його вплив на рак, смертність, старіння, мозок і поведінку, хвороби серця тощо
Ізотіоціанати є одними з найважливіших рослинних сполук, які ви можете отримати у своєму раціоні. У цьому відео я роблю для них найповнішу справу, яку коли-небудь робили. Короткий період уваги? Перейдіть до улюбленої теми, натиснувши один із моментів часу нижче. Повна хронологія нижче.
Ключові розділи:
00:01:14 – Рак і смертність
00:19:04 – Старіння
00:26:30 – Мозок і поведінка
00:38:06 – Підсумок
00:40:27 – Доза
Повний графік:
00:00:34 – Представлення сульфорафану, головна тема відео.
00:01:14 – Споживання овочів хрестоцвітних і зниження смертності від усіх причин.
00:02:12 – Ризик раку передміхурової залози.
00:02:23 – Ризик раку сечового міхура.
00:02:34 – Ризик раку легенів у курців.
00:02:48 – Ризик раку молочної залози.
00:03:13 – Гіпотетична: що робити, якщо у вас уже рак? (інтервенційний)
00:03:35 – Імовірний механізм, який керує асоціативними даними раку та смертності.
00:04:38 – Сульфорафан і рак.
00:05:32 – Докази на тваринах, що показують сильний вплив екстракту паростків брокколі на розвиток пухлин сечового міхура у щурів.
00:06:06 – Вплив прямого прийому сульфорафану у пацієнтів з раком передміхурової залози.
00:07:09 – Біоакумуляція метаболітів ізотіоціаната у фактичній тканині молочної залози.
00:08:32 – Пригнічення стовбурових клітин раку молочної залози.
00:08:53 – Урок історії: ще в Стародавньому Римі стверджували, що капустяні гриби мають оздоровчі властивості.
00:09:16 – Здатність сульфорафану посилювати виведення канцерогену (бензолу, акролеїну).
00:09:51 – NRF2 як генетичний перемикач через елементи антиоксидантної реакції.
00:10:10 – Як активація NRF2 посилює виведення канцерогену через глутатіон-S-кон'югати.
00:10:34 – Брюссельська капуста підвищує глутатіон-S-трансферазу і зменшує пошкодження ДНК.
00:11:20 – Напій з проростків брокколі збільшує виведення бензолу на 61%.
00:15:45 – Споживання хрестоцвітних овочів і смертність від серцево-судинних захворювань.
00:16:55 – порошок паростків брокколі покращує рівень ліпідів у крові та загальний ризик серцевих захворювань у діабетиків 2 типу.
00:19:04 – Початок секції старіння.
00:19:21 – Дієта, збагачена сульфорафаном, збільшує тривалість життя жуків від 15 до 30% (за певних умов).
00:20:34 – Важливість слабкого запалення для довголіття.
00:22:05 – Овочі хрестоцвітних і порошок паростків брокколі, здається, зменшують широкий спектр запальних маркерів у людей.
00:23:40 – Підсумок у середині відео: розділи про рак, старіння
00:24:14 – Дослідження на мишах показують, що сульфорафан може покращити адаптивну імунну функцію в літньому віці.
00:25:18 – Сульфорафан покращив ріст волосся у мишачої моделі облисіння. Зображення на 00:26:10.
00:26:30 – Початок розділу «Мозок і поведінка».
00:27:18 – Вплив екстракту паростків брокколі на аутизм.
00:27:48 – Вплив глюкорафаніну на шизофренію.
00:28:17 – Початок обговорення депресії (правдоподібний механізм та дослідження).
00:31:21 – Дослідження на мишах з використанням 10 різних моделей депресії, викликаної стресом, показало, що сульфорафан так само ефективний, як і флуоксетин (прозак).
00:32:00 – Дослідження показує, що пряме вживання глюкорафаніну мишами так само ефективне для запобігання депресії через модель стресу соціальної поразки.
00:33:01 – Початок відділу нейродегенерації.
00:33:30 – Сульфорафан і хвороба Альцгеймера.
00:33:44 – Сульфорафан і хвороба Паркінсона.
00:33:51 – Сульфорафан і хвороба Хантінгтона.
00:34:13 – Сульфорафан збільшує кількість білків теплового шоку.
00:34:43 – Початок секції черепно-мозкової травми.
00:35:01 – Сульфорафан, введений відразу після ЧМТ, покращує пам’ять (дослідження на мишах).
00:35:55 – Сульфорафан і нейрональна пластичність.
00:36:32 – Сульфорафан покращує навчання на моделі діабету ІІ типу у мишей.
00:37:19 – Сульфорафанова і м’язова дистрофія Дюшенна.
00:37:44 – Інгібування міостатину в клітинах-супутниках м’язів (in vitro).
00:38:06 – Пізнє відео: смертність і рак, пошкодження ДНК, окислювальний стрес і запалення, виділення бензолу, серцево-судинні захворювання, діабет ІІ типу, вплив на мозок (депресія, аутизм, шизофренія, нейродегенерація), шлях NRF2.
00:40:27 – Думки щодо визначення дози паростків брокколі або сульфорафану.
00:41:01 – Анекдоти про проростання в домашніх умовах.
00:43:14 – Про температуру приготування та активність сульфорафану.
00:43:45 – Перетворення сульфорафану з глюкорафаніну кишковими бактеріями.
00:44:24 – Добавки працюють краще в поєднанні з активною мирозиназою з овочів.
00:44:56 – Техніка приготування та овочі хрестоцвітних.
Нагрівання зменшує активність білка епітіоспецифічного білка та збільшує утворення сульфорафану в брокколі
абстрактний
Сульфорафан, ізотіоціанат з брокколі, є одним з найпотужніших антиканцерогенів харчового походження. Ця сполука не присутня в неушкодженому овочі, вона утворюється з його попередника глюкозинолату, глюкорафаніну, під дією мірозинази, ферменту тіоглюкозидази, коли тканину брокколі подрібнюють або жують. Проте низка досліджень продемонструвала, що вихід сульфорафану з глюкорафаніну низький, і що небіологічно активний аналог нітрилу, сульфорафаннітрил, є основним продуктом гідролізу при подрібненні рослинної тканини при кімнатній температурі. Останні дані свідчать про те, що у арабідопсису утворення нітрилу з глюкозинолатів контролюється термочутливим білком, епітіоспецифічним білком (ESP), некаталітичним кофактором мірозинази. Наші цілі полягали в тому, щоб вивчити вплив нагрівання суцвіть і паростків брокколі на утворення сульфорафану та сульфорафанового нітрилу, визначити, чи містить брокколі активність ESP, а потім корелювати зміни в активності ESP, вмісту сульфорафану та біоактивності, що залежать від тепла, виміряні індукцією фаза II детоксикаційного ферменту хінонредуктази (QR) у культурі клітин. Нагрівання свіжих суцвіть брокколі або паростків брокколі до 60 °C перед гомогенізацією одночасно збільшує утворення сульфорафану та зменшує утворення сульфорафанового нітрилу. Значна втрата активності ESP паралельно зі зменшенням утворення сульфорафанового нітрилу. Нагрівання до 70 °C і вище зменшувало утворення обох продуктів у суцвіттях брокколі, але не в паростках брокколі. Індукція QR у культивованих клітинах гепатоми Hepa lclc7 миші одночасно збільшує утворення сульфорафану.
Попереднє нагрівання суцвіть і паростків брокколі до 60 °C значно підвищило каталізоване мирозиназою утворення сульфорафану (SF) в екстрактах рослинної тканини після подрібнення. Це було пов’язано зі зниженням утворення сульфорафанового нітрилу (SF Nitrile) та активності епітіоспецифічного білка (ESP).
На закінчення можна сказати, що сульфорафан є фітохімічною речовиною, яка міститься в брокколі та інших овочах хрестоцвітних. Неконтрольована кількість окислювачів, викликана як внутрішніми, так і зовнішніми факторами, може викликати окислювальний стрес в організмі людини, що в кінцевому підсумку може призвести до різноманітних проблем зі здоров’ям. Сульфорафан може активувати вироблення Nrf2, фактора транскрипції, який допомагає регулювати захисні антиоксидантні механізми, які контролюють реакцію клітини на окислювачі. Обсяг нашої інформації обмежено хіропрактикою та проблемами здоров’я хребта. Щоб обговорити тему, зверніться до доктора Хіменеса або зв’яжіться з нами за адресою�915-850-0900.
Додаткова тема для обговорення: «Гострий біль у спині».
Біль у спині� є однією з найпоширеніших причин інвалідності та пропущених робочих днів у всьому світі. Біль у спині є другою за поширеністю причиною відвідувань лікаря, переважаючи лише інфекції верхніх дихальних шляхів. Приблизно 80 відсотків населення відчувають біль у спині хоча б раз у житті. Хребет – це складна структура, що складається з кісток, суглобів, зв’язок і м’язів, серед інших м’яких тканин. Через це травми та/або загострення стану, такі як�грижі диски, може зрештою призвести до симптомів болю в спині. Спортивні травми або травми в автомобільній катастрофі часто є найчастішою причиною болю в спині, однак іноді найпростіші рухи можуть мати хворобливі наслідки. На щастя, альтернативні варіанти лікування, такі як хіропрактика, можуть допомогти полегшити біль у спині за допомогою корекції хребта та ручних маніпуляцій, що в кінцевому підсумку покращує полегшення болю.
Як правило, окислювачі виробляються контрольованим способом, щоб регулювати основні процеси в організмі людини, включаючи поділ клітин, запалення, імунну функцію, аутофагію та реакцію на стрес. Однак неконтрольоване виробництво цих окислювачів може сприяти окислювальний стрес, що може вплинути на функцію клітин, що призведе до розвитку токсичності, хронічних захворювань та раку. Захисні антиоксидантні механізми людського організму регулюються серією життєво важливих шляхів, які контролюють реакцію клітини на окислювачі. Фактор, пов’язаний з ядерним фактором еритроїду 2, інакше відомий як Nrf2, є новим регулятором стійкості клітин до окисників. Метою статті нижче є обговорення та демонстрація нової ролі Nrf2 у функції мітохондрій.
абстрактний
Фактор транскрипції NF-E2 p45-пов'язаний фактор 2 (Nrf2; назва гена NFE2L2) дозволяє адаптуватися та виживати в умовах стресу, регулюючи експресію генів різноманітних мереж цитопротекторних білків, включаючи антиоксидантні, протизапальні та детоксикаційні ферменти. як білки, які допомагають у відновленні або видаленні пошкоджених макромолекул. Nrf2 відіграє вирішальну роль у підтримці клітинного окислювально-відновного гомеостазу, регулюючи біосинтез, використання та регенерацію глутатіону, тіоредоксину та НАДФН, а також контролюючи виробництво активних форм кисню мітохондріями та НАДФН-оксидазою. У гомеостатичних умовах Nrf2 впливає на потенціал мітохондріальної мембрани, окислення жирних кислот, доступність субстратів (NADH і FADH2/сукцинат) для дихання та синтез АТФ. В умовах стресу або стимуляції фактором росту активація Nrf2 протидіє збільшеному виробленню активних форм кисню в мітохондріях через посилення транскрипції білка 3, що роз’єднує, і впливає на біогенез мітохондрій, підтримуючи рівні ядерного респіраторного фактора 1 і рецептора, активованого проліфератором пероксисом? коактиватора 1?, а також шляхом сприяння біосинтезу пуринових нуклеотидів. Фармакологічні активатори Nrf2, такі як природний ізотіоціанат сульфорафан, інгібують опосередковане окислювачем відкриття мітохондріальної проникної пори та набряк мітохондрій. Цікаво, що синтетична сполука 1,4-дифеніл-1,2,3-триазолу, спочатку розроблена як активатор Nrf2, сприяє мітофагії, тим самим сприяючи загальному мітохондріальному гомеостазу. Таким чином, Nrf2 є помітним гравцем у підтримці структурної та функціональної цілісності мітохондрій, і ця роль є особливо важливою в умовах стресу.
Ключові слова:Біоенергетика, цитозахист, Keap1, мітохондрії, Nrf2, вільні радикали
мелірування
Nrf2 відіграє вирішальну роль у підтримці клітинного окислювально-відновного гомеостазу.
Nrf2 впливає на потенціал мітохондріальної мембрани і синтез АТФ.
Nrf2 впливає на окислення мітохондріальних жирних кислот.
Nrf2 підтримує структурну та функціональну цілісність мітохондрій.
Активатори Nrf2 мають сприятливий вплив, коли функція мітохондрій порушена.
Вступ
Фактор транскрипції NF-E2 p45-пов’язаний фактор 2 (Nrf2; назва гена NFE2L2) регулює експресію мереж генів, що кодують білки з різноманітною цитопротекторною активністю. Сам Nrf2 контролюється насамперед на рівні стабільності білка. У базальних умовах Nrf2 є короткоживучим білком, який піддається безперервному убіквітинації та протеасомній деградації. Існують три відомі убіквітин-лігазні системи, які сприяють деградації Nrf2. Історично, першим негативним регулятором Nrf2, який був відкритий, був Kelch-подібний ECH-асоційований білок 1 (Keap1) [1], протеїн-адаптер субстрату для убіквітин-лігази Cullin 3 (Cul3)/Rbx1 [2], [3], [ 4]. Keap1 використовує високоефективний циклічний механізм для націлювання на Nrf2 для убіквітинування та протеасомної деградації, під час якого Keap1 безперервно регенерується, що дозволяє циклу продовжуватися (рис. 1A) [5]. Nrf2 також піддається деградації, опосередкованої глікогенсинтаз-кіназою (GSK)3/?-TrCP-залежною убіквітин-лігазою на основі Cul1 [6], [7]. Зовсім нещодавно повідомлялося, що в умовах стресу ендоплазматичної мережі Nrf2 убіквітинується та розкладається в процесі, опосередкованому убіквітин-лігазою E3 Hrd1 [8].
Рисунок 1 Модель циклічного послідовного зв'язування та регенерації для Keap1-опосередкованої деградації Nrf2. (A) Nrf2 зв'язується послідовно з вільним димером Keap1: спочатку через свій високоафінний домен зв'язування ETGE (червоні палички), а потім через його низькоафінний домен зв'язування DLG (чорні палички). У цій конформації білкового комплексу Nrf2 піддається убіквітинації і націлений на протеасомну деградацію. Вільний Keap1 відновлюється і здатний зв’язуватися з щойно перекладеним Nrf2, і цикл починається знову. (B) Індуктори (білі ромби) реагують з сенсорними цистеїнами Keap1 (сині палички), що призводить до конформаційних змін і порушення активності адаптера субстрату. Вільний Keap1 не регенерується, а знову синтезований Nrf2 накопичується і переміщається в ядро.
На додаток до того, що Keap1 служить білком-адаптером субстрату убіквітин-лігази, Keap2 також є датчиком для широкого спектру низькомолекулярних активаторів Nrf9 (іменованих індукторами) [1]. Індуктори блокують цикл Keap2-опосередкованої деградації Nrf1 шляхом хімічної модифікації специфічних залишків цистеїну в Keap10 [11], [1] або шляхом безпосереднього порушення межі зв’язування Keap2:Nrf12 [13], [2]. Отже, Nrf1 не руйнується, а транскрипційний фактор накопичується і переміщається в ядро (рис. 14В), де він утворює гетеродимер з невеликим білком Maf; зв’язується з елементами антиоксидантної реакції, верхніми регуляторними областями своїх цільових генів; і ініціює транскрипцію [15], [16], [2]. Батарея мішеней NrfXNUMX містить білки з різноманітними цитопротекторними функціями, включаючи ферменти метаболізму ксенобіотиків, білки з антиоксидантними та протизапальними функціями, протеасомальні субодиниці, а також білки, які регулюють клітинний окислювально-відновний гомеостаз і беруть участь у проміжному метаболізмі.
Nrf2: головний регулятор клітинного окислювально-відновного гомеостазу
Функція Nrf2 як головного регулятора клітинного окислювально-відновного гомеостазу широко визнана. Експресія генів як каталітичної, так і регуляторної субодиниць ?-глутамілцистеїн-лігази, ферменту, що каталізує етап, що обмежує швидкість біосинтезу відновленого глутатіону (GSH), безпосередньо регулюється Nrf2 [17]. Субодиниця xCT системи xc-, яка імпортує цистин у клітини, також є прямою мішенню транскрипції Nrf2 [18]. У клітині цистин зазнає перетворення в цистеїн, попередник для біосинтезу GSH. На додаток до своєї ролі в біосинтезі GSH, Nrf2 забезпечує засоби для підтримки глутатіону в його відновленому стані за допомогою скоординованої регуляції транскрипції глутатіонредуктази 1 [19], [20], яка відновлює окислений глутатіон до GSH за допомогою відновних еквівалентів з NADPH. . Необхідний НАДФН забезпечується чотирма основними ферментами, що утворюють НАДФН, яблучним ферментом 1 (ME1), ізоцитратдегідрогеназою 1 (IDH1), глюкозо-6-фосфатдегідрогеназою (G6PD) і 6-фосфоглюконатдегідрогеназою (PGD). транскрипційно регулюється частково Nrf2 (рис. 2) [21], [22], [23], [24]. Цікаво, що Nrf2 також регулює індукційну експресію генів цитозольних, мікросомальних і мітохондріальних форм альдегіддегідрогенази [25], які використовують NAD(P)+ як кофактор, викликаючи NAD(P)H. Дійсно, рівні NADPH і співвідношення NADPH/NADP+ нижчі в ембріональних фібробластах, виділених з Nrf2-нокаутних (Nrf2-KO) мишей, порівняно з клітинами їх побратимів дикого типу (WT), а рівні NADPH зменшуються після нокдауну Nrf2 в лінії ракових клітин з конститутивно активним Nrf2 [26]. Як і очікувалося, рівні GSH нижчі в клітинах, в яких Nrf2 був порушений; навпаки, активація Nrf2 генетичними або фармакологічними засобами призводить до підвищення GSH [27], [28], [29]. Важливо, що Nrf2 також регулює експресію генів тіоредоксину [30], [31], [32], тіоредоксинредуктази 1 [28], [29], [32], [33] та сульфіредоксину [34], які є важливими для відновлення окислених білкових тіолів.
Рисунок 2 Роль Nrf2 в метаболізмі швидко проліферуючих клітин. Nrf2 є позитивним регулятором генів, що кодують ферменти як окислювальної групи [тобто глюкозо-6-фосфатдегідрогенази (G6PD) і 6-фосфоглюконатдегідрогенази (PGD)], так і неокислювальної групи [тобто трансальдолаза 1 (TALDO1) і транскетолаза ( TKT)] пентозофосфатного шляху. G6PD і PGD генерують NADPH. Nrf2 також регулює експресію генів двох інших ферментів, що утворюють НАДФН, яблучного ферменту 1 (ME1) та ізоцитратдегідрогенази 1 (IDH1). Експресія генів фосфорибозилпірофосфат амідотрансферази (PPAT), яка каталізує вступ у шлях біосинтезу пуринів de novo, також позитивно регулюється Nrf2, так само як і експресія метилентетрагідрофолатдегідрогенази 2 (MTHFD2), яка виконує критичну роль у мітохондріях. забезпечення одновуглецевих одиниць для біосинтезу пуринів de novo. Піруваткіназа (PK) негативно регулюється Nrf2 і, як очікується, сприятиме накопиченню гліколітичних проміжних продуктів і, разом з G6PD, каналізації метаболітів через пентозофосфатний шлях і синтезу нуклеїнових кислот, амінокислот і фосфоліпідів. Nrf2 негативно регулює експресію гена АТФ-цитрат-ліази (CL), що може збільшити доступність цитрату для мітохондріальної утилізації або (через ізоцитрат) для IDH1. Червоний і синій вказують на позитивну і негативну регуляцію відповідно. Мітохондрія показана сірим кольором. Скорочення метаболітів: G-6-P, глюкозо-6-фосфат; F-6-P, фруктозо-6-фосфат; F-1,6-BP, фруктозо-1,6-біфосфат; GA-3-P, гліцеральдегід 3-фосфат; 3-PG, 3-фосфогліцерат; PEP, фосфоенолпіруват; 6-P-Gl, 6-фосфоглюконолактон; 6-PG, 6-фосфоглюконат; R-5-P, рибулозо-5-фосфат; PRPP, 5-фосфорибозил-?-1-пірофосфат; ТГФ, тетрагідрофолат; IMP, інозинмонофосфат; АМФ, аденозинмонофосфат; GMP, гуанозинмонофосфат.
Враховуючи вирішальну роль Nrf2 як головного регулятора клітинного окислювально-відновного гомеостазу, не дивно, що порівняно з клітинами WT рівні активних форм кисню (ROS) вищі в клітинах, в яких Nrf2 був порушений (Nrf2-KO). [35]. Ця різниця особливо помітна при зараженні агентами, що викликають окислювальний стрес. Крім того, клітини з дефіцитом Nrf2 набагато чутливіші до токсичності окислювачів різних типів і не можуть бути захищені індукторами Nrf2, які за тих же умов забезпечують ефективний і тривалий захист клітин WT [29], [36] , [37]. На додаток до загального клітинного окислювально-відновного гомеостазу, Nrf2 також має вирішальне значення для підтримки мітохондріального окислювально-відновного гомеостазу. Таким чином, порівняно з WT, загальний мітохондріальний пул NADH значно збільшується в Keap1-KO і різко зменшується в клітинах Nrf2-KO [35].
За допомогою візуалізації живих клітин ми нещодавно відстежували швидкість виробництва АФК у первинних гліонейрональних кокультурах і зрізах тканини мозку, виділених від мишей WT, Nrf2-KO або Keap1-нокдаун (Keap1-KD) [38]. Як і очікувалося, швидкість виробництва АФК була швидшою в клітинах і тканинах Nrf2-KO в порівнянні з їхніми аналогами WT. Проте ми зробили несподіване спостереження, що порівняно з WT клітини Keap1-KD також мають вищі показники продукції АФК, хоча величина різниці між генотипами WT та Keap1-KD була меншою, ніж між WT та Nrf2-KO. . Потім ми проаналізували рівні мРНК NOX2 і NOX4, каталітичних субодиниць двох ізоформ NADPH оксидази (NOX), які були причетні до патології мозку, і виявили, що NOX2 різко збільшується в умовах дефіциту Nrf2, тоді як NOX4 посилюється, коли Nrf2 є конститутивно активованим, хоча і в меншій мірі. У кількісному відношенні величина підвищення регуляції в клітинах і тканинах від мутантних мишей співпадає з відповідним збільшенням продукції АФК [38]. Цікаво, що Nrf2 не тільки регулює НАДФН-оксидазу, а й АФК, що виробляється НАДФН-оксидазою, може активувати Nrf2, як показано в епітеліальних клітинах легенів і кардіоміоцитах [39], [40]. Крім того, зовсім недавнє дослідження продемонструвало, що НАДФН-оксидаза-залежна активація Nrf2 є важливим ендогенним механізмом захисту від пошкодження мітохондрій і загибелі клітин у серці під час хронічного перевантаження тиском [41].
На додаток до каталітичної активності НАДФН-оксидази, мітохондріальне дихання є ще одним основним внутрішньоклітинним джерелом АФК. За допомогою мітохондрій-специфічного зонда MitoSOX ми дослідили внесок АФК мітохондріального походження в загальну продукцію АФК у первинних ізольованих гліонейрональних культурах. від мишей WT, Nrf2-KO або Keap1-KD [38]. Як і очікувалося, клітини Nrf2-KO мали вищі показники виробництва мітохондріальних АФК, ніж WT. Відповідно до висновків щодо загального виробництва АФК, показники виробництва мітохондріальних АФК у Keap1-KD також були вищими порівняно з клітинами WT. Важливо, що блокування комплексу I з ротеноном викликало різке збільшення виробництва АФК в мітохондріях як в клітинах WT, так і в Keap1-KD, але не впливало на клітини Nrf2-KO. На відміну від очікуваного збільшення мітохондріальної продукції АФК в клітинах WT після додавання пірувату (для підвищення доступності NADH, збільшення потенціалу мітохондріальної мембрани та нормалізації дихання), продукція АФК зменшилася в клітинах Nrf2-KO. Разом ці висновки свідчать про те, що за відсутності Nrf2: (i) активність комплексу I порушується, (ii) порушення активності комплексу I пов’язане з обмеженням субстратів, і (iii) порушення активності комплексу I є однією з основних причин збільшення мітохондріальної продукції АФК, можливо, через зворотний потік електронів з комплексу II.
Nrf2 впливає на потенціал мітохондріальної мембрани та дихання
Потенціал мітохондріальної мембрани (??m) є універсальним індикатором здоров’я мітохондрій і метаболічного стану клітини. У здоровій клітині ??m підтримується мітохондріальним дихальним ланцюгом. Цікаво, що стабільне ізотопне мічення амінокислотами в культуральному протеомічному дослідженні в лінії клітин MCF10A естрогенного епітелію грудної залози з негативним рецептором естрогену показало, що компонент мітохондріального ланцюга транспорту електронів NDUFA4 посилюється фармакологічною активацією (за допомогою сульфорафану, Nrroforphane). тоді як генетичне підвищення регуляції Nrf2 (за допомогою нокдауну Keap2) призводить до зниження регуляції субодиниць цитохром с оксидази COX1 і COX2I4 [1]. Дослідження протеома печінки за допомогою двовимірного гель-електрофорезу та матричної лазерної десорбції/іонізаційної мас-спектрометрії виявило, що Nrf42 регулює експресію субодиниці АТФ-синтази ? [2]. Крім того, повідомляється, що мітохондріальний білок DJ-43, який відіграє роль у підтримці активності комплексу I [1], стабілізує Nrf44 [2], [45], хоча нейропротекторні ефекти фармакологічної або генетичної активації Nrf46 не залежать від DJ-2 [1]. Однак наслідки цих спостережень для функції мітохондрій не були досліджені.
Відповідно до порушення активності комплексу I в умовах дефіциту Nrf2, базальний β?m нижчий у ембріональних фібробластах (MEFs) миші Nrf2-KO і культивованих первинних гліонейрональних клітинах порівняно з їхніми аналогами WT (рис. 3, вставка) [35]. Навпаки, базальний ??m вищий, коли Nrf2 генетично конститутивно регулюється (за допомогою нокдауну або нокауту Keap1). Ці відмінності в ??m між генотипами вказують на те, що на дихання впливає активність Nrf2. Дійсно, оцінка споживання кисню в базальному стані показала, що порівняно з WT споживання кисню нижче у МЕФ Nrf2-KO та Keap1-KO на ~50 та ~35 % відповідно.
Рисунок 3 Запропонований механізм порушення функції мітохондрій в умовах дефіциту Nrf2. (1) Зниження рівнів ME1, IDH1, G6PD і PGD призводить до зниження рівня NADPH. (2) Рівні GSH також низькі. (3) Низька активність ME1 може зменшити пул пірувату, що надходить у мітохондрії. (4) Генерація NADH відбувається повільніше, що призводить до порушення активності комплексу I і збільшення продукції АФК мітохондріями. (5) Відновлення FAD до FADH2 в мітохондріальних білках також зменшується, знижуючи потік електронів від FADH2 до UbQ і в комплекс III. (6) Повільніше утворення UbQH2 може знизити активність ферменту сукцинатдегідрогенази. (7) Підвищені рівні АФК можуть додатково інгібувати активність комплексу II. (8) Нижча ефективність окислення жирних кислот сприяє зниженню доступності субстрату для мітохондріального дихання. (9) Гліколіз посилюється як компенсаторний механізм для зниження виробництва АТФ при окисному фосфорилюванні. (10) АТФ-синтаза діє у зворотному порядку, щоб підтримувати ??m. Червоний і синій вказують на підвищення та зниження регуляції відповідно. Коробки означають наявність експериментальних доказів. На вставці показані зображення мітохондрій кортикальних астроцитів WT і Nrf2-KO, візуалізовані за допомогою потенціометричного флуоресцентного зонда метилового ефіру тетраметилродаміну (TMRM; 25 нМ). Масштабна смуга, 20 м.
Ці відмінності в ??m і диханні між генотипами відображаються швидкістю використання субстратів для мітохондріального дихання. Застосування субстратів для циклу трикарбонової кислоти (TCA) (малат/піруват, які, у свою чергу, збільшують продукцію субстрату комплексу I NADH) або метилсукцинату, субстрату для комплексу II, спричиняє ступінчасте збільшення ??m в обох WT і нейрони Keap1-KD, але швидкість збільшення вища в клітинах Keap1-KD. Що ще важливіше, форми відповіді на ці субстрати циклу TCA відрізняються між двома генотипами, завдяки чому швидке підвищення ??m в клітинах Keap1-KD після додавання субстрату супроводжується швидким падінням, а не плато, що свідчить про незвичайну швидке споживання субстрату. Ці результати тісно узгоджуються з набагато нижчими (на 50%) рівнями малату, пірувату і сукцинату, які спостерігалися після 70-годинного імпульсу [U-1C13]глюкози в Keap6-KO в порівнянні з WT MEF. клітини [1]. У нейронах Nrf24-KO тільки піруват здатний збільшувати β?m, тоді як малат і метилсукцинат викликають помірну деполяризацію. Вплив Nrf2 на продукцію мітохондріального субстрату, здається, є основним механізмом, за допомогою якого Nrf2 впливає на функцію мітохондрій. Редокс-індекс мітохондріального NADH (баланс між споживанням NADH комплексом I та продукцією NADPH у циклі TCA) значно нижчий у клітинах Nrf2-KO порівняно з їхніми аналогами WT, а також швидкість регенерації пулів NADH і FADH2 після інгібування комплексу IV (за допомогою NaCN) діють повільніше в мутантних клітинах.
У мітохондріях, ізольованих з мозку та печінки мишей, додавання субстратів для комплексу I або для комплексу II сильніше збільшує швидкість споживання кисню, коли Nrf2 активується, і менш ефективно, коли Nrf2 порушується [35]. Таким чином, малат викликає вищу швидкість споживання кисню в Keap1-KD порівняно з WT, але його ефект слабший в мітохондріях Nrf2-KO. Аналогічно, у присутності ротенону (коли комплекс I інгібується), сукцинат активує споживання кисню більшою мірою в Keap1-KD порівняно з WT, тоді як відповідь у мітохондріях Nrf2-KO зменшується. Крім того, первинні нейрональні культури Nrf2-KO та миші є більш чутливими до токсичності інгібіторів комплексу II 3-нітропропіонової кислоти та малонату, тоді як внутрішньостриарна трансплантація астроцитів, що надекспресують Nrf2, є захисною [48], [49]. Аналогічно, миші Nrf2-KO більш чутливі до нейротоксичності, спричиненої інгібітором комплексу I 2-метил-1-фенілпіридинію в іоні 4-метил-1-феніл-4, тоді як генетична або фармакологічна активація Nrf1,2,3,6 має захисний ефект. 49-тетрагідропіридинова модель хвороби Паркінсона на тваринах [50], [51], [52], [53], [54], [55], [56], [57], [58], [59], [60], [61], [XNUMX].
Коефіцієнт респіраторного контролю (RCR), відношення стану 3 (стимульований ADP) до стану 4 (відсутня ADP), зменшується за відсутності Nrf2, але RCR подібний між мітохондріями Keap1-KD і WT [35 ]. Оскільки RCR є показником ступеня зв’язку активності мітохондріального дихального ланцюга з окисним фосфорилюванням, цей висновок вказує на те, що вища швидкість дихання в мітохондріях Keap1-KD не пов’язана з роз’єднанням окисного фосфорилювання. Це також свідчить про те, що окисне фосфорилювання є більш ефективним, коли Nrf2 активований. Вища швидкість дихання в мітохондріях Keap1-KD узгоджується з вищими рівнями виробництва мітохондріальних АФК [38], оскільки більша швидкість дихання може призвести до збільшення витоку електронів. Однак в умовах окисного стресу підвищеній продукції АФК протидіє Nrf2-залежна транскрипційна регуляція роз’єднувального білка 3 (UCP3), яка підвищує протонну провідність внутрішньої мембрани мітохондрій і, отже, зменшує продукцію супероксиду [62]. Зовсім недавно було показано, що продукт перекисного окислення ліпідів 4-гідрокси-2-ноненал опосередковує Nrf2-залежну регуляцію UCP3 в кардіоміоцитах; це може бути особливо важливим для захисту в умовах окисного стресу, наприклад під час реперфузії ішемії [63].
Nrf2 впливає на ефективність окисного фосфорилювання та синтез АТФ
Відповідно до впливу Nrf2 на дихання, у мітохондріях мозку та печінки, дефіцит Nrf2 призводить до зниження ефективності окисного фосфорилювання (за оцінкою відношення АДФ до кисню, який витрачається на синтез АТФ), тоді як активація Nrf2 (Keap1 -KD) має протилежний ефект [35]. Порівняно з WT, рівні АТФ значно вищі в клітинах з конститутивною регуляцією Nrf2 і нижчі, коли Nrf2 знищений [64] або порушений [35]. Крім того, використання інгібіторів окисного фосфорилювання (олігоміцин) або гліколізу (йодооцтова кислота) показало, що Nrf2 змінює спосіб, за допомогою якого клітини виробляють АТФ. Таким чином, в нейронах WT олігоміцин викликає повне падіння АТФ і йодооцтова кислота не має подальшого впливу. Примітно, що в клітинах Nrf2-KO олігоміцин підвищує рівень АТФ, який потім повільно, але повністю виснажується йодооцтовою кислотою, що вказує на те, що за відсутності Nrf2 основним джерелом виробництва АТФ є гліколіз, а не окисне фосфорилювання. Цікаво, що незважаючи на підвищену ефективність окисного фосфорилювання в клітинах Keap1-KD, додавання олігоміцину призводить до ~80% зниження рівня АТФ, а йодооцтова кислота викликає подальше зниження на ~20%. Таким чином, або дефіцит Nrf2, або його конститутивна активація зменшує внесок окисного фосфорилювання і збільшує внесок гліколізу в синтез АТФ. Цей ефект особливо виражений, коли Nrf2 відсутній і узгоджується із залежністю ??m від присутності глюкози в середовищі [35] і підвищеними рівнями гліколітичних проміжних продуктів (G-6-P, F-6-P , дигідроксиацетонфосфат, піруват і лактат) після нокдауну Nrf2 [24].
Підвищення рівня АТФ після інгібування F1F0-АТФази олігоміцином вказує на те, що за відсутності Nrf2 F1F0-АТФаза функціонує як АТФаза, а не АТФ-синтаза, тобто діє у зворотному порядку. Така зміна активності, швидше за все, відображає необхідність перекачування протонів через внутрішню мітохондріальну мембрану в спробі зберегти ??m, що має вирішальне значення для функціональної цілісності цієї органели. Про зміну функції F1F0-АТФази також свідчить спостережувана деполяризація мітохондрій при введенні олігоміцину в клітини Nrf2-KO, що різко контрастує з гіперполяризацією, що виникає в їхніх WT або Keap1-дефіцитних аналогах [35]. Загалом, схоже, що в умовах дефіциту Nrf2 АТФ виробляється в основному в результаті гліколізу, і цей АТФ потім частково використовується F1F0-АТФазою для підтримки ??m.
Вплив дефіциту Nrf2 на ??m особливо виражений, коли клітини інкубують у середовищі без глюкози, а ??m є на ~50% нижчим у Nrf2-KO порівняно з клітинами WT [35]. В умовах нестачі глюкози окислення мітохондріальних жирних кислот (FAO) є основним постачальником субстратів для дихання та окисного фосфорилювання, що свідчить про те, що Nrf2 може впливати на FAO. Справді, ефективність FAO як для довголанцюгової (C16:0) насиченої жирної кислоти пальмітинової кислоти, так і для коротколанцюгової (C6:0) гексанової кислоти вища в Keap1-KO MEFs та ізольованих мітохондріях серця і печінки, ніж у їх WT аналогів, тоді як він нижчий у клітинах Nrf2-KO та мітохондріях [65]. Ці ефекти також мають велике значення для людей: дійсно, метаболічні зміни, що вказують на кращу інтеграцію FAO з активністю циклу TCA, як повідомлялося, відбуваються в дослідженнях людського втручання з дієтами, багатими на глюкорафанін, попередник класичного активатора Nrf2 сульфорафану [ 66].
Під час першого етапу мітохондріальної FAO про-R водень ?-вуглецю виходить у вигляді гідриду, який відновлює кофактор FAD до FADH2, який, у свою чергу, передає електрони на убіхінон (UbQ) у дихальному ланцюгу, що в кінцевому підсумку сприяє виробленню АТФ. . У той час як стимуляція FAO пальмітоілкарнітином за відсутності глюкози викликає очікуване підвищення рівня АТФ в клітинах WT і Keap1-KO, при цьому зростання АТФ відбувається швидше в клітинах Keap1-KO, ідентична обробка не викликає змін АТФ в Nrf2-KO. MEFs [65]. Цей експеримент демонструє, що за відсутності Nrf2 ФАО пригнічується, і, крім того, це передбачає придушення ФАО як одну з причин зниження рівня АТФ в умовах дефіциту Nrf2 [35], [64].
Примітно, що людські 293 Т-клітини, в яких Nrf2 був заглушений, мають нижчу експресію CPT1 і CPT2[67], двох ізоформ карнітинпальмітоілтрансферази (CPT), ферменту, що обмежує швидкість в мітохондріальних FAO. Відповідно, рівні мРНК Cpt1 нижчі в печінці Nrf2-KO порівняно з мишами WT [68]. CPT каталізує перенесення ацильної групи довголанцюгового жирного ацил-КоА від коензиму А до L-карнітину і таким чином дозволяє імпортувати ацилкарнітин з цитоплазми в мітохондрії. Хоча це не було досліджено на сьогоднішній день, можливо, що на додаток до транскрипційного впливу на експресію CPT1, Nrf2 також може впливати на функцію цього ферменту, контролюючи рівні його основного алостеричного інгібітора, малоніл-КоА. Це пояснюється тим, що за механізмом, який наразі неясний, Nrf2 негативно регулює експресію стеароіл-КоА-десатурази (SCD) [69] і цитрат-ліази (CL) [69], [70]. Цікаво, що нокаут або інгібування SCD призводить до посилення фосфорилювання та активації AMP-активованої протеїнкінази (AMPK) [71], [72], [73], і можна припустити, що за відсутності Nrf2 рівні SCD підвищиться, у свою чергу, знизить активність AMPK. Це може бути додатково посилено зниженими рівнями білка AMPK, які спостерігалися в печінці мишей Nrf2-KO [68], що тісно узгоджується з підвищеними рівнями AMPK, про які повідомлялося в печінці Keap1-KD. миші [74]. Одним із наслідків зниженої активності AMPK є полегшення його інгібуючого фосфорилювання (на Ser79) ацетил-КоА карбоксилази (ACC) [75], яке може бути додатково підвищене транскрипції за відсутності Nrf2, оскільки воно зменшується активацією Nrf2 [70]. ]. Висока активність ACC у поєднанні з підвищеною експресією CL, що збільшує вироблення ацетил-КоА, субстрату для ACC, може в кінцевому підсумку підвищити рівень продукту ACC, малоніл-КоА. Високі рівні малоніл-КоА інгібують CPT, тим самим зменшуючи транспорт жирних кислот у мітохондрії. Нарешті, Nrf2 позитивно регулює експресію CD36 [76], транслокази, яка імпортує жирні кислоти через плазматичні та мітохондріальні мембрани. Таким чином, одним з механізмів, за допомогою якого Nrf2 може впливати на ефективність мітохондріальної FAO, є регулювання імпорту довголанцюгових жирних кислот в мітохондрії.
На додаток до прямої регуляції транскрипції, Nrf2 також може змінювати ефективність мітохондріальної FAO через його вплив на клітинний окислювально-відновний метаболізм. Це може бути особливо актуальним, коли активність Nrf2 низька або відсутня, умови, які зміщують клітинний окислювально-відновний статус у бік окисленого стану. Дійсно, кілька ферментів ФАО було ідентифіковано як чутливі до окислювально-відновних змін. Одним з таких ферментів є дуже довголанцюгова ацил-КоА-дегідрогеназа (VLCAD), яка сприяє більш ніж 80% активності дегідрування пальмітоїл-КоА в тканинах людини [77]. Цікаво, що Hurd et al. [78] показали, що VLCAD містить залишки цистеїну, які значно змінюють свій окислювально-відновний стан під час впливу H2O2 на ізольовані мітохондрії серця щура. Крім того, S-нітрозилування VLCAD печінки мишей на Cys238 покращує каталітичну ефективність ферменту [79], і цілком ймовірно, що окислення того ж цистеїну може мати протилежний ефект, зрештою знижуючи ефективність мітохондріальної FAO. Таким чином, можливо, що, хоча рівні експресії VLCAD істотно не відрізняються в МЕФ WT, Nrf2-KO або Keap1-KO [65], активність ферменту VLCAD може бути нижчою за відсутності Nrf2 через більш високі рівні. ROS.
На основі всіх цих висновків можна припустити, що (рис. 3): за відсутності Nrf2 рівні NADPH нижчі через зниження експресії ME1, IDH1, G6PD та PGD. Рівні відновленого глутатіону також нижчі через зниження експресії ферментів, які беруть участь у його біосинтезі та регенерації, та нижчих рівнів NADPH, які необхідні для перетворення окисленої у відновлену форму глутатіону. Низька експресія ME1 зменшить пул пірувату, що надходить у мітохондрії, при цьому гліколіз стане основним джерелом пірувату. Генерація NADH відбувається повільніше, що призводить до порушення активності комплексу I і збільшення продукції АФК мітохондріями. Відновлення FAD до FADH2 також повільніше, принаймні частково через менш ефективне окислення жирних кислот, що порушує потік електронів від FADH2 до UbQ і в комплекс III. Оскільки UbQH2 є активатором сукцинатдегідрогенази [80], уповільнення його утворення може знизити активність ферменту сукцинатдегідрогенази. Підвищені рівні супероксиду та перекису водню можуть додатково інгібувати активність комплексу II [81]. Нижча ефективність окислення жирних кислот сприяє зниженню доступності субстрату для мітохондріального дихання та виробництва АТФ при окисному фосфорилюванні. Як компенсаторний механізм посилюється гліколіз. АТФ-синтаза функціонує у зворотному порядку, як АТФаза, намагаючись підтримувати ??m.
Nrf2 і мітохондріальний біогенез
Повідомлялося, що порівняно з WT печінка мишей Nrf2-KO має нижчий вміст мітохондрій (що визначається співвідношенням мітохондріальної та ядерної ДНК); це ще більше зменшується 24-годинним голодуванням у мишей WT і Nrf2-KO; навпаки, хоча й не відрізняється від WT за нормальних умов годування, вміст мітохондрій у мишей з високою активністю Nrf2 не впливає на голодування [82]. Цікаво, що добавка активатора Nrf2 (R)-?-ліпоєвої кислоти [83], [84], [85] сприяє мітохондріальному біогенезу в адипоцитах 3T3-L1 [86]. Два класи ядерних регуляторів транскрипції відіграють вирішальну роль у мітохондріальному біогенезі. До першого класу належать фактори транскрипції, такі як ядерні респіраторні фактори11 і 2, які контролюють експресію генів, що кодують субодиниці п’яти дихальних комплексів, мітохондріальних трансляційних компонентів і біосинтетичних ферментів гему, які локалізовані в мітохондріальному матриксі [88]. Piantadosi та ін. [89] показали, що Nrf2-залежна транскрипційна регуляція ядерного респіраторного фактора 1 сприяє мітохондріальному біогенезу та захищає від цитотоксичності кардіотоксичного антрациклінового хіміотерапевтичного засобу доксорубіцину. На противагу цьому, Zhang et al. [82] повідомили, що генетична активація Nrf2 не впливає на експресію базальної мРНК ядерного респіраторного фактора 1 у печінці мишей.
Другим класом ядерних регуляторів транскрипції з критичними функціями в мітохондріальному біогенезі є коактиватори транскрипції, такі як рецептор, активований проліфератором пероксисом ? коактиватори (PGC)1? і 1?, які взаємодіють з факторами транскрипції, механізмами базальної транскрипції та РНК-сплайсингу, а також ферментами, що модифікують гістон [88], [90], [91]. На експресію сімейства коактиваторів PGC1 впливають численні сигнали навколишнього середовища. Обробка фібробластів людини активатором Nrf2 сульфорафаном викликає збільшення маси мітохондрій та індукцію PGC1? і PGC1? [92], хоча потенційна залежність від Nrf2 у цьому дослідженні не досліджувалася. Однак миші з цукровим діабетом, у яких Nrf2 або активований гіпоморфним нокдауном гена Keap1 (db/db:Keap1flox/?:Nrf2+/+), або порушений (db/db:Keap1flox/?:Nrf2?/?), мають нижчий печінковий PGC1? рівні експресії, ніж у контрольних тварин (db/db:Keap1flox/+:Nrf2+/+) [93]. Немає відмінностей у рівнях мРНК для PGC1? спостерігаються в печінці недіабетичних мишей, які мають або WT, або Nrf2-KO, тоді як ці рівні нижчі у тварин з надмірною експресією Nrf2 (Keap1-KD і специфічний для печінки Keap1-KO) [82]. Примітно, що 24-годинне голодування підвищує рівень PGC1? мРНК у печінці мишей усіх генотипів, але збільшення значно більше в печінці Nrf2-KO порівняно з мишами з надекспресією WT або Nrf2. Порівняно з WT, миші Nrf2-KO, у яких зазнала септична інфекція або гостре ураження легень через інфекцію, демонструють ослаблену транскрипційну регуляцію ядерного респіраторного фактора 1 і PGC1? [94], [95]. Разом ці спостереження свідчать про те, що роль Nrf2 у підтримці рівнів як ядерного респіраторного фактора 1, так і PGC1? є складним і стає найбільш помітним в умовах стресу.
Крім експресії генів, що кодують мітохондріальні білки, біогенез мітохондрій вимагає синтезу нуклеотидів. Генетична активація Nrf2 посилює біосинтез пуринів шляхом активізації пентозофосфатного шляху та метаболізму фолатів і глутаміну, особливо в клітинах, що швидко проліферують (рис. 2) [24]. Аналіз транскриптому мутантної дрозофіли з дефіцитом мітохондріальної серин/треонін протеїнкінази PTEN-індукованої передбачуваної кінази 1 (PINK1) показав, що мітохондріальна дисфункція призводить до активізації транскрипції генів, що впливають на метаболізм нуклеотидів, що свідчить про посилення біонуклеотидів [96]. являє собою механізм захисту від нейротоксичних наслідків дефіциту PINK1. Nrf2 регулює експресію фосфорибозилпірофосфат амідотрансферази (PPAT), яка каталізує входження в шлях біосинтезу пуринових нуклеотидів de novo, і мітохондріальної метилентетрагідрофолатдегідрогенази 2 (MTHFD2) (рис. 2). Останній є біфункціональним ферментом з активністю дегідрогенази та циклогідролази, який має вирішальне значення для забезпечення як гліцину, так і формиату як джерела одновуглецевих одиниць для біосинтезу пуринів у швидкозростаючих клітинах [97]. Тому імовірно, що активація Nrf2 може бути захисною і може повернути мітохондріальну дисфункцію при дефіциті PINK1. Дійсно, фармакологічна активація Nrf2 сульфорафаном або тритерпеноїдом RTA-408 відновлює ??m і захищає PINK1-дефіцитні клітини від токсичності дофаміну [98]. Хоча основні механізми здаються складними, разом ці дані вказують на те, що активність Nrf2 може впливати на біогенез мітохондрій, впливаючи на рівні експресії критичних транскрипційних факторів і коактиваторів, а також шляхом посилення біосинтезу нуклеотидів.
Nrf2 і цілісність мітохондрій
Хоча прямі докази не завжди доступні, є серйозні ознаки того, що Nrf2 важливий для цілісності мітохондрій, особливо в умовах окисного стресу. Мітохондрії, виділені з мозку та печінки щурів, яким ввели одноразову дозу сульфорафану-активатора Nrf2, стійкі до відкриття пори переходу проникності мітохондрій (mPTP), викликаного окислювачем трет-бутилгідропероксидом [99], [100]. Нещодавно було виявлено, що mPTP, комплекс, який дозволяє внутрішній мембрані мітохондрій стати проникною для молекул з масою до 1500 Да, утворюється з димерів F0F1-АТФ-синтази [101]. Опосередкована сульфорафаном резистентність до відкриття mPTP корелює з підвищеним антиоксидантним захистом, а рівні мітохондріального GSH, глутатіонпероксидази 1, яблучного ферменту 3 і тіоредоксину 2 підвищуються у фракціях мітохондрій, виділених у тварин, які отримували сульфорафан [100].
Пошкодження мітохондріальних білків і порушення дихання, викликані продуктом електрофільного перекисного окислення ліпідів 4-гідрокси-2-ноненаль, ослаблені в мітохондріях, виділених з кори головного мозку мишей, які отримували сульфорафан [102]. В епітеліальних клітинах нирок щурів і в нирках сульфорафан захищає від токсичності, спричиненої цисплатином і гентаміцином, і втрати β?m[103], [104]. Захист від панелі окислювачів (супероксид, перекис водню, пероксинітрит) і електрофілів (4-гідрокси-2-ноненал і акролеїн) і підвищення мітохондріального антиоксидантного захисту також спостерігали при обробці гладком’язових клітин аорти щурів сульфорафаном [105]. ]. У моделі гострого ураження нирок, індукованого контрастом, нещодавно було показано, що ішемічне прекондиціонування кінцівок має захисні ефекти, включаючи інгібування відкриття mPTP і набряку мітохондрій шляхом активації Nrf2 внаслідок інгібування GSK3? [106].
Мітофагія, процес, за допомогою якого дисфункціональні мітохондрії вибірково поглинаються аутофагосомами і доставляються в лізосоми для деградації та перероблення клітиною, має важливе значення для мітохондріального гомеостазу [107], [108]. Хоча причинний зв’язок між Nrf2 та мітофагією не встановлено, є докази того, що фактор транскрипції може бути важливим у контролі якості мітохондрій, відіграючи певну роль у мітофагії. Це може бути особливо помітним в умовах окисного стресу. Так, у моделі сепсису підвищення рівнів маркера аутофагосоми MAP1 легкого ланцюга 3-II (LC3-II) і білка вантажу p62 через 24 години після інфекції пригнічується у Nrf2-KO порівняно з мишами WT [109] . Нещодавно було відкрито маломолекулярний індуктор мітофагії (так званий p62-опосередкований індуктор мітофагії, PMI); ця сполука 1,4-дифеніл-1,2,3-триазолу спочатку була розроблена як активатор Nrf2, який порушує взаємодію фактора транскрипції з Keap1 [110]. Подібно до клітин, в яких Nrf2 генетично регулюється (Keap1-KD або Keap1-KO), клітини, що піддаються впливу PMI, мають більш високий рівень спокою. Важливо, що збільшення мітохондріальної локалізації LC3, яке спостерігається після обробки PMI клітин WT, не відбувається в клітинах Nrf2-KO, що свідчить про залучення Nrf2.
Нарешті, ультраструктурний аналіз зрізів печінки виявив наявність набряклих мітохондрій із зменшеною кристою та зруйнованими мембранами в гепатоцитах Nrf2-KO, але не WT, мишей, яких годували дієтою з високим вмістом жирів протягом 24 тижнів; зокрема, ця печінка демонструє чіткі ознаки окисного стресу та запалення [68]. Можна зробити висновок, що Nrf2 відіграє вирішальну роль у підтримці цілісності мітохондрій в умовах окисного та запального стресу.
Сульфорафан та його вплив на рак, смертність, старіння, мозок і поведінку, хвороби серця тощо
Ізотіоціанати є одними з найважливіших рослинних сполук, які ви можете отримати у своєму раціоні. У цьому відео я роблю для них найповнішу справу, яку коли-небудь робили. Короткий період уваги? Перейдіть до улюбленої теми, натиснувши один із моментів часу нижче. Повна хронологія нижче.
Ключові розділи:
00:01:14 – Рак і смертність
00:19:04 – Старіння
00:26:30 – Мозок і поведінка
00:38:06 – Підсумок
00:40:27 – Доза
Повний графік:
00:00:34 – Представлення сульфорафану, головна тема відео.
00:01:14 – Споживання овочів хрестоцвітних і зниження смертності від усіх причин.
00:02:12 – Ризик раку передміхурової залози.
00:02:23 – Ризик раку сечового міхура.
00:02:34 – Ризик раку легенів у курців.
00:02:48 – Ризик раку молочної залози.
00:03:13 – Гіпотетична: що робити, якщо у вас уже рак? (інтервенційний)
00:03:35 – Імовірний механізм, який керує асоціативними даними раку та смертності.
00:04:38 – Сульфорафан і рак.
00:05:32 – Докази на тваринах, що показують сильний вплив екстракту паростків брокколі на розвиток пухлин сечового міхура у щурів.
00:06:06 – Вплив прямого прийому сульфорафану у пацієнтів з раком передміхурової залози.
00:07:09 – Біоакумуляція метаболітів ізотіоціаната у фактичній тканині молочної залози.
00:08:32 – Пригнічення стовбурових клітин раку молочної залози.
00:08:53 – Урок історії: ще в Стародавньому Римі стверджували, що капустяні гриби мають оздоровчі властивості.
00:09:16 – Здатність сульфорафану посилювати виведення канцерогену (бензолу, акролеїну).
00:09:51 – NRF2 як генетичний перемикач через елементи антиоксидантної реакції.
00:10:10 – Як активація NRF2 посилює виведення канцерогену через глутатіон-S-кон'югати.
00:10:34 – Брюссельська капуста підвищує глутатіон-S-трансферазу і зменшує пошкодження ДНК.
00:11:20 – Напій з проростків брокколі збільшує виведення бензолу на 61%.
00:15:45 – Споживання хрестоцвітних овочів і смертність від серцево-судинних захворювань.
00:16:55 – порошок паростків брокколі покращує рівень ліпідів у крові та загальний ризик серцевих захворювань у діабетиків 2 типу.
00:19:04 – Початок секції старіння.
00:19:21 – Дієта, збагачена сульфорафаном, збільшує тривалість життя жуків від 15 до 30% (за певних умов).
00:20:34 – Важливість слабкого запалення для довголіття.
00:22:05 – Овочі хрестоцвітних і порошок паростків брокколі, здається, зменшують широкий спектр запальних маркерів у людей.
00:23:40 – Підсумок у середині відео: розділи про рак, старіння
00:24:14 – Дослідження на мишах показують, що сульфорафан може покращити адаптивну імунну функцію в літньому віці.
00:25:18 – Сульфорафан покращив ріст волосся у мишачої моделі облисіння. Зображення на 00:26:10.
00:26:30 – Початок розділу «Мозок і поведінка».
00:27:18 – Вплив екстракту паростків брокколі на аутизм.
00:27:48 – Вплив глюкорафаніну на шизофренію.
00:28:17 – Початок обговорення депресії (правдоподібний механізм та дослідження).
00:31:21 – Дослідження на мишах з використанням 10 різних моделей депресії, викликаної стресом, показало, що сульфорафан так само ефективний, як і флуоксетин (прозак).
00:32:00 – Дослідження показує, що пряме вживання глюкорафаніну мишами так само ефективне для запобігання депресії через модель стресу соціальної поразки.
00:33:01 – Початок відділу нейродегенерації.
00:33:30 – Сульфорафан і хвороба Альцгеймера.
00:33:44 – Сульфорафан і хвороба Паркінсона.
00:33:51 – Сульфорафан і хвороба Хантінгтона.
00:34:13 – Сульфорафан збільшує кількість білків теплового шоку.
00:34:43 – Початок секції черепно-мозкової травми.
00:35:01 – Сульфорафан, введений відразу після ЧМТ, покращує пам’ять (дослідження на мишах).
00:35:55 – Сульфорафан і нейрональна пластичність.
00:36:32 – Сульфорафан покращує навчання на моделі діабету ІІ типу у мишей.
00:37:19 – Сульфорафанова і м’язова дистрофія Дюшенна.
00:37:44 – Інгібування міостатину в клітинах-супутниках м’язів (in vitro).
00:38:06 – Пізнє відео: смертність і рак, пошкодження ДНК, окислювальний стрес і запалення, виділення бензолу, серцево-судинні захворювання, діабет ІІ типу, вплив на мозок (депресія, аутизм, шизофренія, нейродегенерація), шлях NRF2.
00:40:27 – Думки щодо визначення дози паростків брокколі або сульфорафану.
00:41:01 – Анекдоти про проростання в домашніх умовах.
00:43:14 – Про температуру приготування та активність сульфорафану.
00:43:45 – Перетворення сульфорафану з глюкорафаніну кишковими бактеріями.
00:44:24 – Добавки працюють краще в поєднанні з активною мирозиназою з овочів.
00:44:56 – Техніка приготування та овочі хрестоцвітних.
00:46:06 – Ізотіоціанати як зоб.
Nrf2 є фактором транскрипції, який відіграє важливу роль у системі клітинного антиоксидантного захисту організму людини. Елемент, що реагує на антиоксиданти, або ARE, є регуляторним механізмом генів. Багато досліджень продемонстрували, що Nrf2 або фактор 2, пов’язаний з NF-E2, регулює широкий спектр генів, керованих ARE, у кількох типах клітин. Було також виявлено, що Nrf2 відіграє важливу роль у захисті клітин та антиканцерогенності, що демонструє, що Nrf2 може бути ефективним засобом лікування нейродегенеративних захворювань та раку, які, як вважають, спричинені окислювальним стресом. Д-р Алекс Хіменес, округ Колумбія, CCST Insight
Заключні зауваження
Хоча багато питань все ще залишаються відкритими, наявні експериментальні дані чітко вказують на те, що Nrf2 є важливим гравцем у підтримці мітохондріального гомеостазу та структурної цілісності. Ця роль стає особливо важливою в умовах окисного, електрофільного та запального стресу, коли здатність посилювати опосередковані Nrf2 цитопротекторні реакції впливає на загальне здоров’я та виживання клітини та організму. Роль Nrf2 у функції мітохондрій являє собою ще один шар широких цитопротекторних механізмів, оркестрованих цим фактором транскрипції. Оскільки багато патологічних станів людини мають окислювальний стрес, запалення та мітохондріальну дисфункцію як важливі компоненти їх патогенезу, фармакологічна активація Nrf2 є перспективною для профілактики та лікування захворювань. Всебічне розуміння точних механізмів, за допомогою яких Nrf2 впливає на функцію мітохондрій, є важливим для раціонального планування майбутніх клінічних досліджень і може запропонувати нові біомаркери для моніторингу терапевтичної ефективності.
Метою статті вище було обговорити, а також продемонструвати роль Nrf2 у функції мітохондрій. Nrf2, або ядерний фактор, пов'язаний з еритроїдом 2, є новим регулятором стійкості клітин до окисників, які можуть сприяти окислювальному стресу, впливаючи на клітинну функцію і призводячи до розвитку токсичності, хронічних захворювань і навіть раку. Хоча виробництво окислювачів в організмі людини може служити для різних цілей, включаючи поділ клітин, запалення, імунну функцію, аутофагію та реакцію на стрес, важливо контролювати їх перевиробництво, щоб запобігти проблемам зі здоров’ям. Обсяг нашої інформації обмежено хіропрактикою та проблемами здоров’я хребта. Щоб обговорити тему, зверніться до доктора Хіменеса або зв’яжіться з нами за адресою�915-850-0900.
Додаткова тема для обговорення: «Гострий біль у спині».
Біль у спині� є однією з найпоширеніших причин інвалідності та пропущених робочих днів у всьому світі. Біль у спині є другою за поширеністю причиною відвідувань лікаря, переважаючи лише інфекції верхніх дихальних шляхів. Приблизно 80 відсотків населення відчувають біль у спині хоча б раз у житті. Хребет – це складна структура, що складається з кісток, суглобів, зв’язок і м’язів, серед інших м’яких тканин. Через це травми та/або загострення стану, такі як�грижі диски, може зрештою призвести до симптомів болю в спині. Спортивні травми або травми в автомобільній катастрофі часто є найчастішою причиною болю в спині, однак іноді найпростіші рухи можуть мати хворобливі наслідки. На щастя, альтернативні варіанти лікування, такі як хіропрактика, можуть допомогти полегшити біль у спині за допомогою корекції хребта та ручних маніпуляцій, що в кінцевому підсумку покращує полегшення болю. �
Інструмент Find A Practitioner від IFM — це найбільша мережа рефералів у функціональній медицині, створена, щоб допомогти пацієнтам знайти практикуючих функціональних лікарів у будь-якій точці світу. Сертифіковані практикуючі лікарі IFM вказані першими в результатах пошуку, враховуючи їхню широку освіту в галузі функціональної медицини