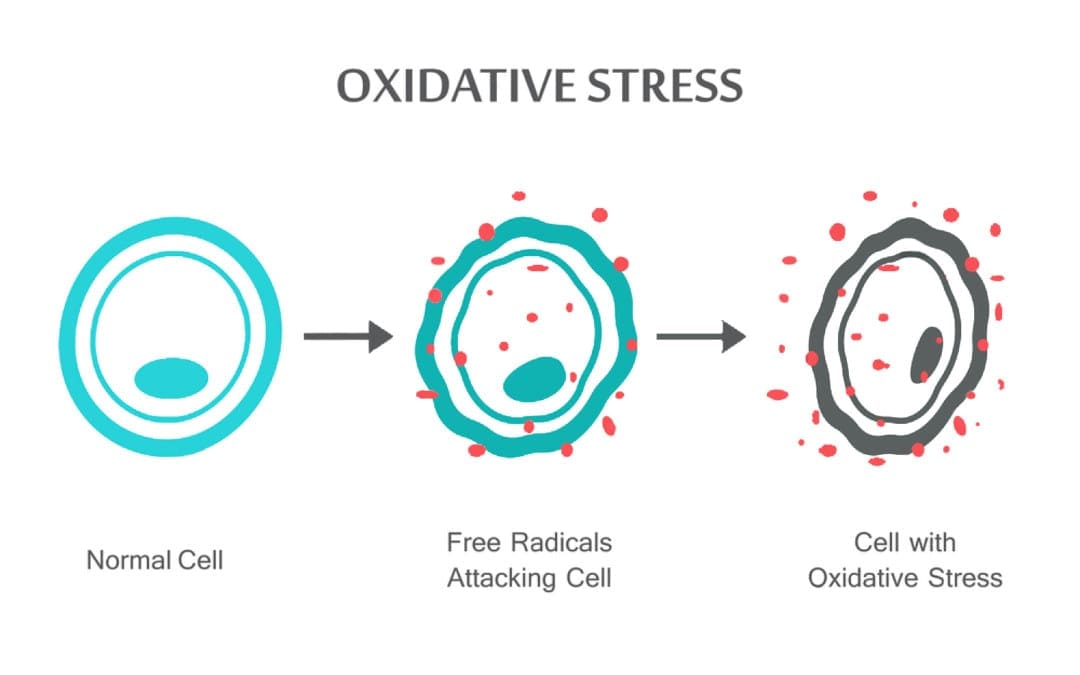
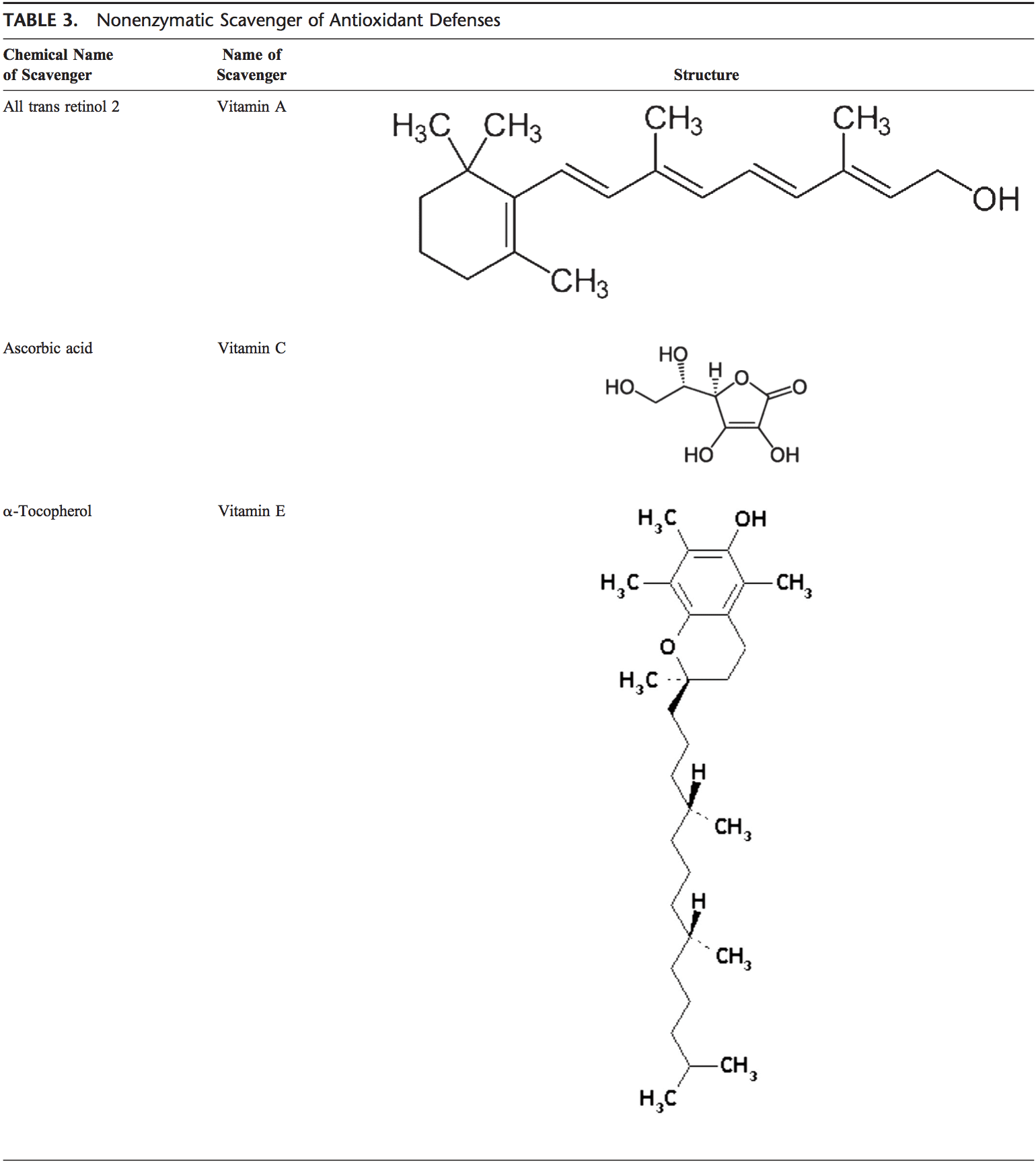
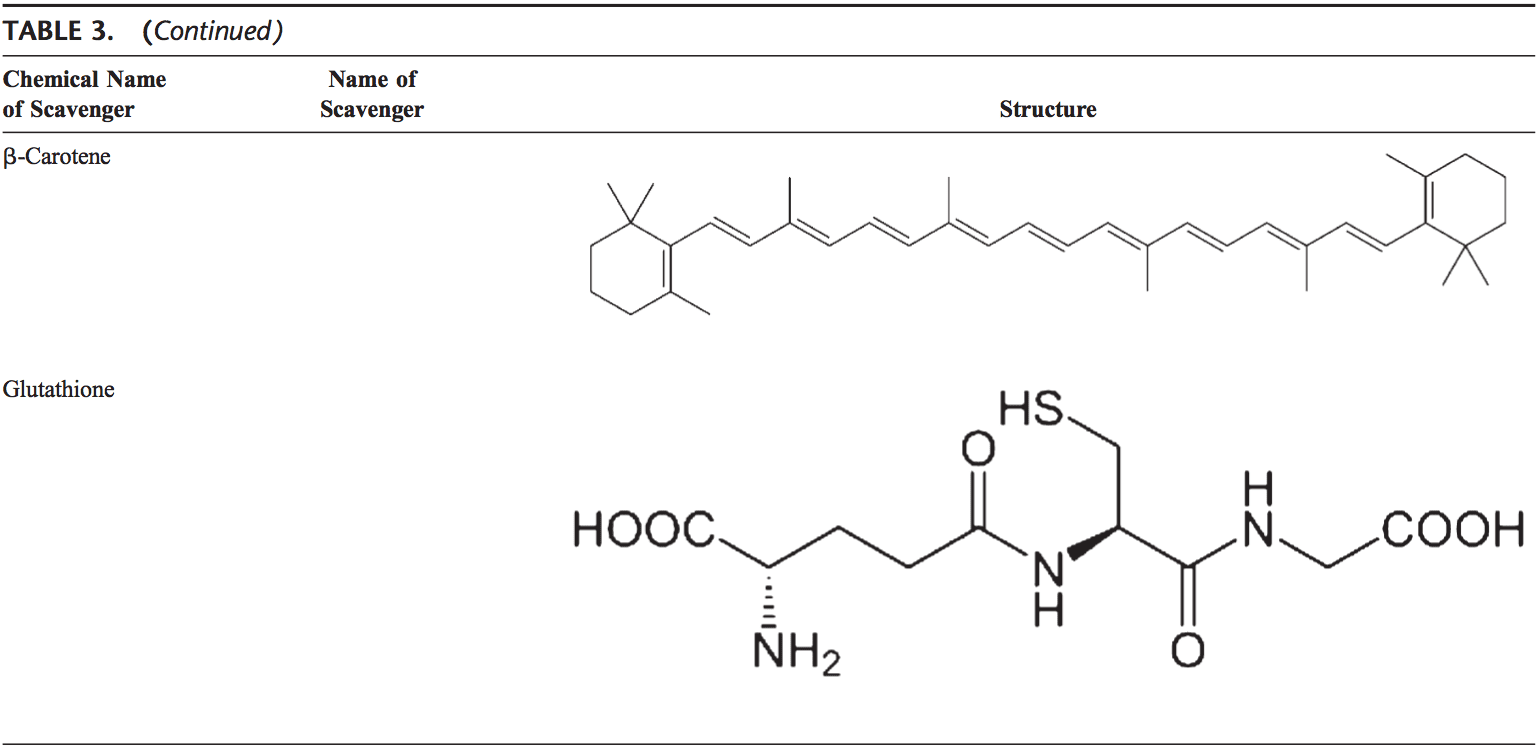
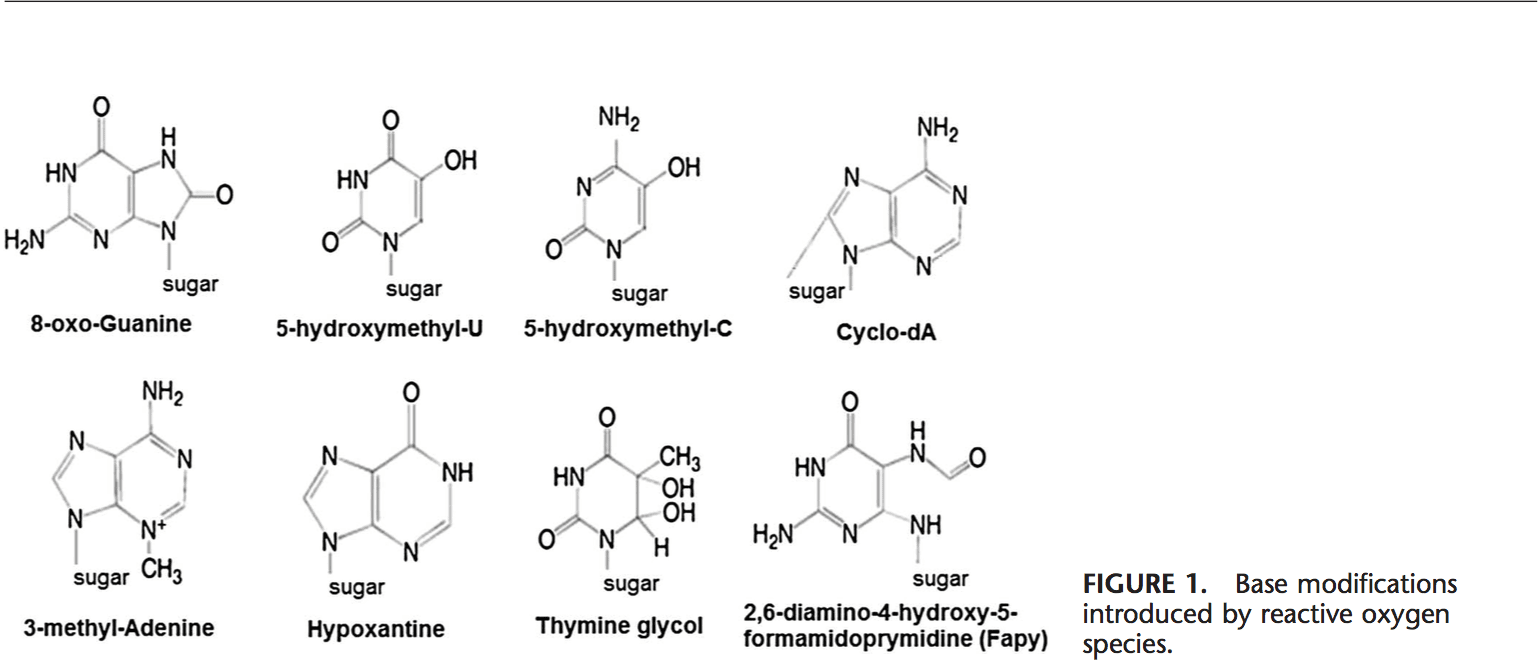
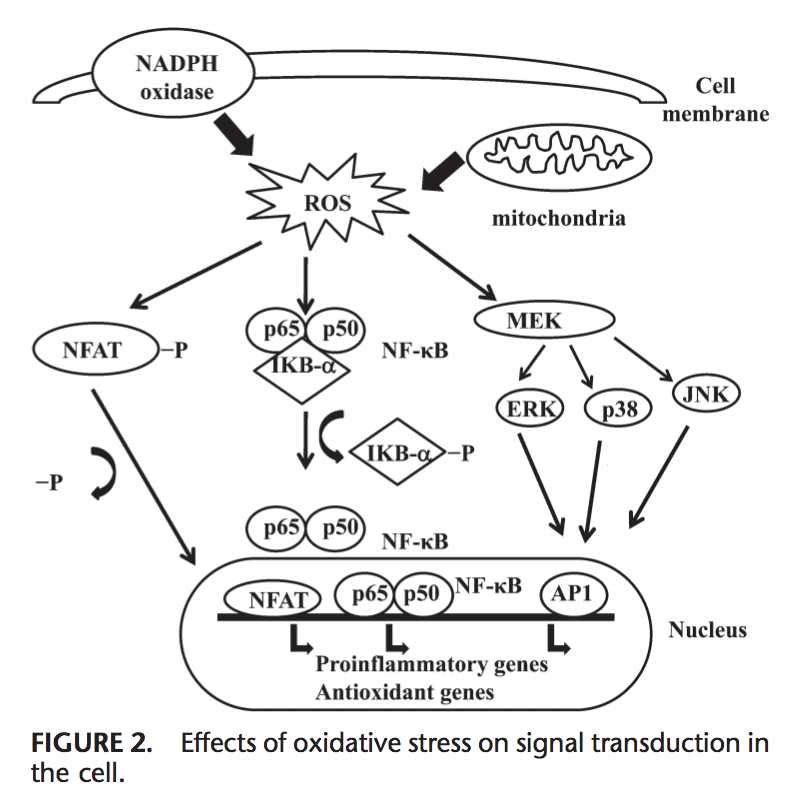
Команда хіропрактики та функціональної медицини в клініці оксидативного стресу. Окислювальний стрес визначається як порушення балансу між виробленням активного кисню (вільних радикалів) і антиоксидантним захистом. Іншими словами, це дисбаланс між виробленням вільних радикалів і здатністю організму протидіяти чи детоксикувати шкідливим впливам шляхом нейтралізації антиоксидантами. Окислювальний стрес призводить до багатьох патофізіологічних станів в організмі. Сюди входять нейродегенеративні захворювання, тобто хвороба Паркінсона, хвороба Альцгеймера, генні мутації, рак, синдром хронічної втоми, синдром крихкої X, захворювання серця та кровоносних судин, атеросклероз, серцева недостатність, серцевий напад та запальні захворювання. Окислення відбувається за кількох обставин:
клітини використовують глюкозу для виробництва енергії
імунна система бореться з бактеріями і викликає запалення
організм детоксикує забруднювачі, пестициди та сигаретний дим
У нашому тілі в будь-який момент часу відбуваються мільйони процесів, які можуть призвести до окислення. Ось кілька симптомів:
Втома
Втрата пам'яті або туман мозку
Біль у м’язах або суглобах
Зморшки разом із сивиною
Зниження зору
Головні болі і чутливість до шуму
Сприйнятливість до інфекцій
Вибір органічних продуктів і уникнення токсинів у вашому оточенні має велике значення. Це, поряд зі зменшенням стресу, може бути корисним для зменшення окислення.
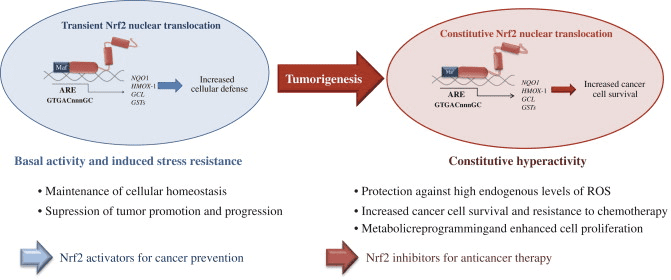
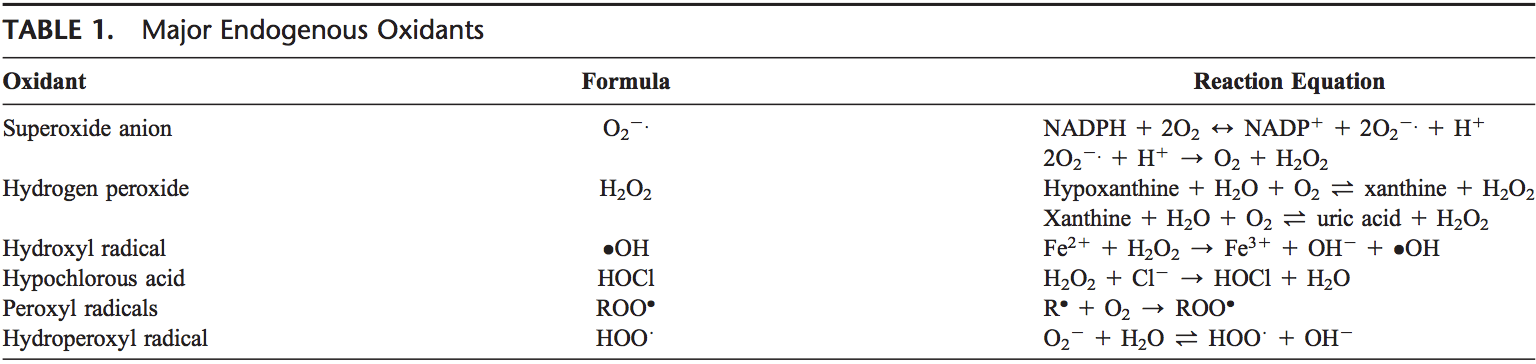
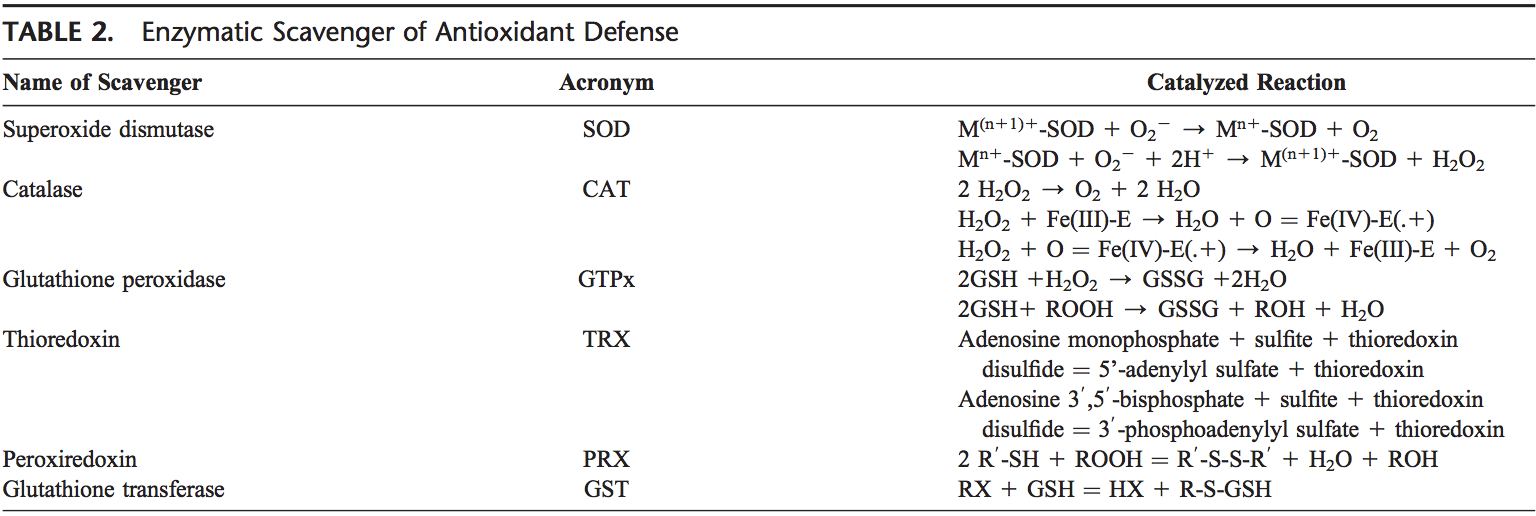
Як правило, окислювачі виробляються контрольованим способом, щоб регулювати основні процеси в організмі людини, включаючи поділ клітин, запалення, імунну функцію, аутофагію та реакцію на стрес. Однак неконтрольоване виробництво цих окислювачів може сприяти окислювальний стрес, що може вплинути на функцію клітин, що призведе до розвитку токсичності, хронічних захворювань та раку. Захисні антиоксидантні механізми людського організму регулюються серією життєво важливих шляхів, які контролюють реакцію клітини на окислювачі. Фактор, пов’язаний з ядерним фактором еритроїду 2, інакше відомий як Nrf2, є новим регулятором стійкості клітин до окисників. Метою статті нижче є обговорення та демонстрація нової ролі Nrf2 у функції мітохондрій.
абстрактний
Фактор транскрипції NF-E2 p45-пов'язаний фактор 2 (Nrf2; назва гена NFE2L2) дозволяє адаптуватися та виживати в умовах стресу, регулюючи експресію генів різноманітних мереж цитопротекторних білків, включаючи антиоксидантні, протизапальні та детоксикаційні ферменти. як білки, які допомагають у відновленні або видаленні пошкоджених макромолекул. Nrf2 відіграє вирішальну роль у підтримці клітинного окислювально-відновного гомеостазу, регулюючи біосинтез, використання та регенерацію глутатіону, тіоредоксину та НАДФН, а також контролюючи виробництво активних форм кисню мітохондріями та НАДФН-оксидазою. У гомеостатичних умовах Nrf2 впливає на потенціал мітохондріальної мембрани, окислення жирних кислот, доступність субстратів (NADH і FADH2/сукцинат) для дихання та синтез АТФ. В умовах стресу або стимуляції фактором росту активація Nrf2 протидіє збільшеному виробленню активних форм кисню в мітохондріях через посилення транскрипції білка 3, що роз’єднує, і впливає на біогенез мітохондрій, підтримуючи рівні ядерного респіраторного фактора 1 і рецептора, активованого проліфератором пероксисом? коактиватора 1?, а також шляхом сприяння біосинтезу пуринових нуклеотидів. Фармакологічні активатори Nrf2, такі як природний ізотіоціанат сульфорафан, інгібують опосередковане окислювачем відкриття мітохондріальної проникної пори та набряк мітохондрій. Цікаво, що синтетична сполука 1,4-дифеніл-1,2,3-триазолу, спочатку розроблена як активатор Nrf2, сприяє мітофагії, тим самим сприяючи загальному мітохондріальному гомеостазу. Таким чином, Nrf2 є помітним гравцем у підтримці структурної та функціональної цілісності мітохондрій, і ця роль є особливо важливою в умовах стресу.
Ключові слова:Біоенергетика, цитозахист, Keap1, мітохондрії, Nrf2, вільні радикали
мелірування
Nrf2 відіграє вирішальну роль у підтримці клітинного окислювально-відновного гомеостазу.
Nrf2 впливає на потенціал мітохондріальної мембрани і синтез АТФ.
Nrf2 впливає на окислення мітохондріальних жирних кислот.
Nrf2 підтримує структурну та функціональну цілісність мітохондрій.
Активатори Nrf2 мають сприятливий вплив, коли функція мітохондрій порушена.
Вступ
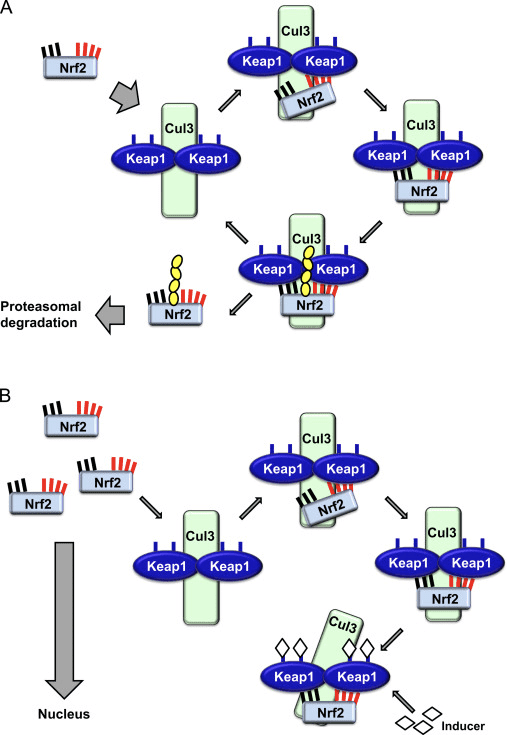
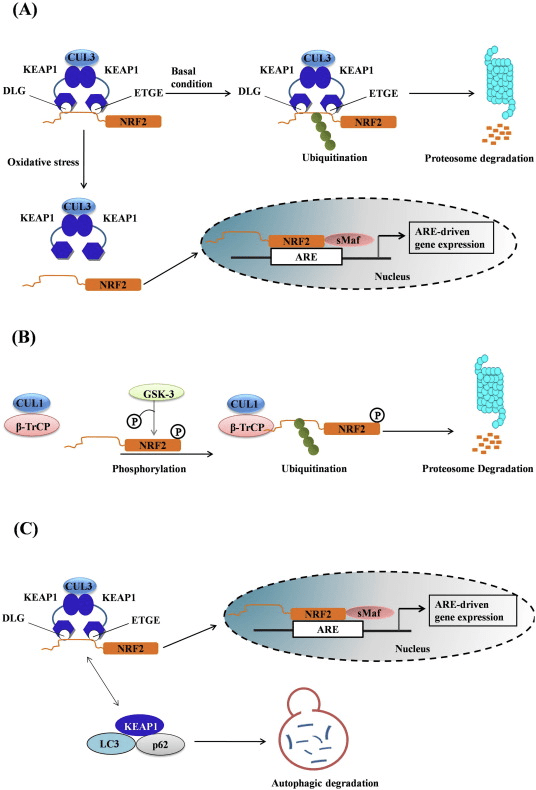
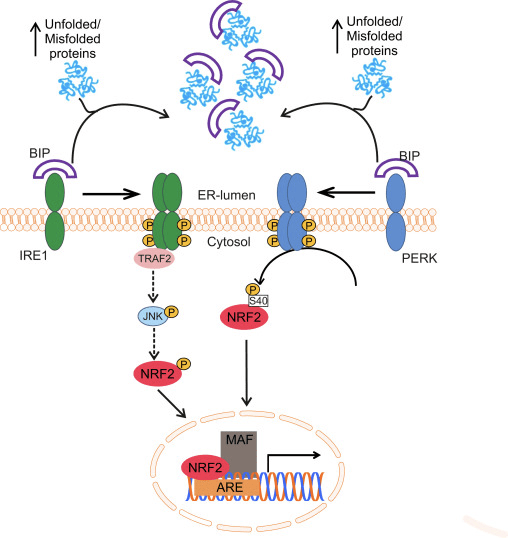
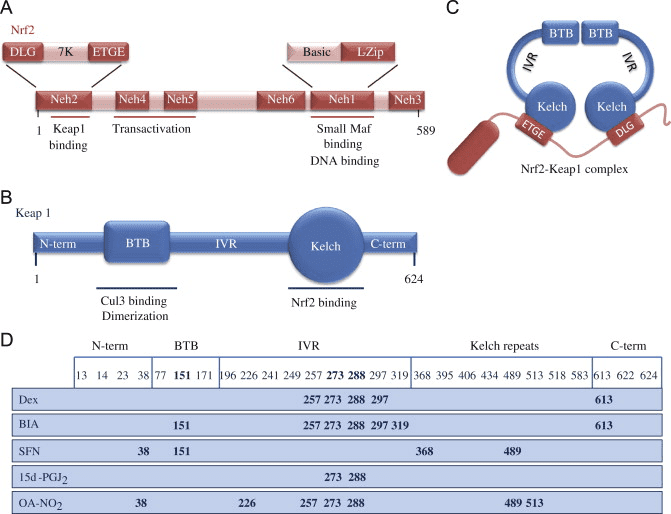
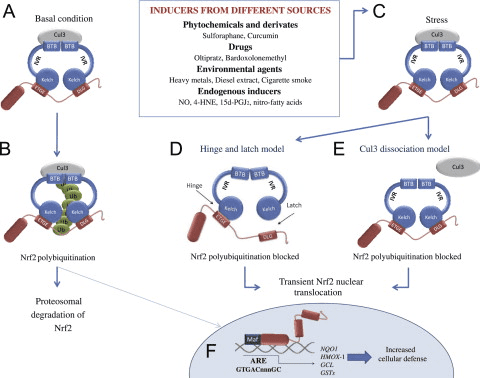
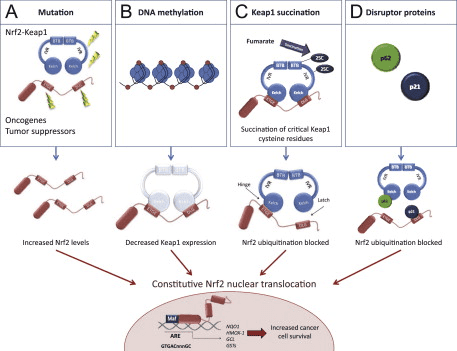
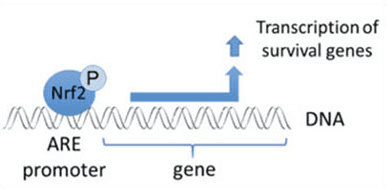
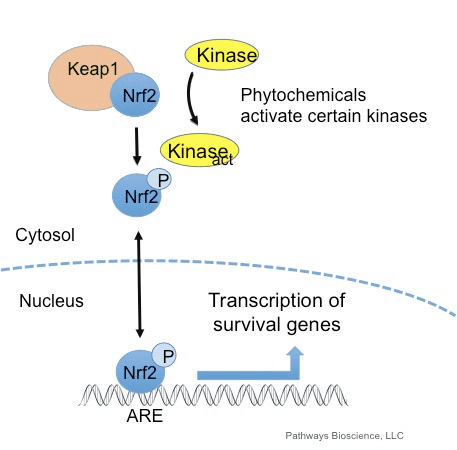
Фактор транскрипції NF-E2 p45-пов’язаний фактор 2 (Nrf2; назва гена NFE2L2) регулює експресію мереж генів, що кодують білки з різноманітною цитопротекторною активністю. Сам Nrf2 контролюється насамперед на рівні стабільності білка. У базальних умовах Nrf2 є короткоживучим білком, який піддається безперервному убіквітинації та протеасомній деградації. Існують три відомі убіквітин-лігазні системи, які сприяють деградації Nrf2. Історично, першим негативним регулятором Nrf2, який був відкритий, був Kelch-подібний ECH-асоційований білок 1 (Keap1) [1], протеїн-адаптер субстрату для убіквітин-лігази Cullin 3 (Cul3)/Rbx1 [2], [3], [ 4]. Keap1 використовує високоефективний циклічний механізм для націлювання на Nrf2 для убіквітинування та протеасомної деградації, під час якого Keap1 безперервно регенерується, що дозволяє циклу продовжуватися (рис. 1A) [5]. Nrf2 також піддається деградації, опосередкованої глікогенсинтаз-кіназою (GSK)3/?-TrCP-залежною убіквітин-лігазою на основі Cul1 [6], [7]. Зовсім нещодавно повідомлялося, що в умовах стресу ендоплазматичної мережі Nrf2 убіквітинується та розкладається в процесі, опосередкованому убіквітин-лігазою E3 Hrd1 [8].
Рисунок 1 Модель циклічного послідовного зв'язування та регенерації для Keap1-опосередкованої деградації Nrf2. (A) Nrf2 зв'язується послідовно з вільним димером Keap1: спочатку через свій високоафінний домен зв'язування ETGE (червоні палички), а потім через його низькоафінний домен зв'язування DLG (чорні палички). У цій конформації білкового комплексу Nrf2 піддається убіквітинації і націлений на протеасомну деградацію. Вільний Keap1 відновлюється і здатний зв’язуватися з щойно перекладеним Nrf2, і цикл починається знову. (B) Індуктори (білі ромби) реагують з сенсорними цистеїнами Keap1 (сині палички), що призводить до конформаційних змін і порушення активності адаптера субстрату. Вільний Keap1 не регенерується, а знову синтезований Nrf2 накопичується і переміщається в ядро.
На додаток до того, що Keap1 служить білком-адаптером субстрату убіквітин-лігази, Keap2 також є датчиком для широкого спектру низькомолекулярних активаторів Nrf9 (іменованих індукторами) [1]. Індуктори блокують цикл Keap2-опосередкованої деградації Nrf1 шляхом хімічної модифікації специфічних залишків цистеїну в Keap10 [11], [1] або шляхом безпосереднього порушення межі зв’язування Keap2:Nrf12 [13], [2]. Отже, Nrf1 не руйнується, а транскрипційний фактор накопичується і переміщається в ядро (рис. 14В), де він утворює гетеродимер з невеликим білком Maf; зв’язується з елементами антиоксидантної реакції, верхніми регуляторними областями своїх цільових генів; і ініціює транскрипцію [15], [16], [2]. Батарея мішеней NrfXNUMX містить білки з різноманітними цитопротекторними функціями, включаючи ферменти метаболізму ксенобіотиків, білки з антиоксидантними та протизапальними функціями, протеасомальні субодиниці, а також білки, які регулюють клітинний окислювально-відновний гомеостаз і беруть участь у проміжному метаболізмі.
Nrf2: головний регулятор клітинного окислювально-відновного гомеостазу
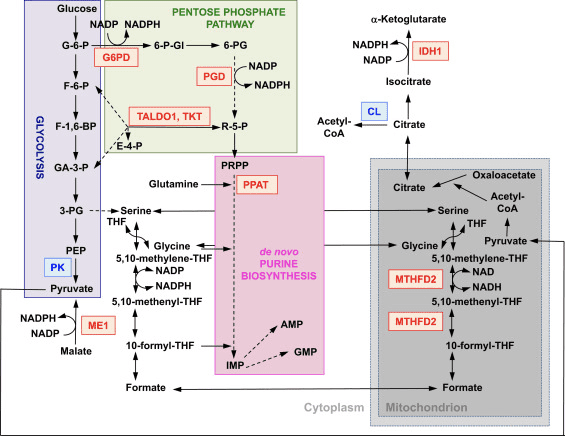
Функція Nrf2 як головного регулятора клітинного окислювально-відновного гомеостазу широко визнана. Експресія генів як каталітичної, так і регуляторної субодиниць ?-глутамілцистеїн-лігази, ферменту, що каталізує етап, що обмежує швидкість біосинтезу відновленого глутатіону (GSH), безпосередньо регулюється Nrf2 [17]. Субодиниця xCT системи xc-, яка імпортує цистин у клітини, також є прямою мішенню транскрипції Nrf2 [18]. У клітині цистин зазнає перетворення в цистеїн, попередник для біосинтезу GSH. На додаток до своєї ролі в біосинтезі GSH, Nrf2 забезпечує засоби для підтримки глутатіону в його відновленому стані за допомогою скоординованої регуляції транскрипції глутатіонредуктази 1 [19], [20], яка відновлює окислений глутатіон до GSH за допомогою відновних еквівалентів з NADPH. . Необхідний НАДФН забезпечується чотирма основними ферментами, що утворюють НАДФН, яблучним ферментом 1 (ME1), ізоцитратдегідрогеназою 1 (IDH1), глюкозо-6-фосфатдегідрогеназою (G6PD) і 6-фосфоглюконатдегідрогеназою (PGD). транскрипційно регулюється частково Nrf2 (рис. 2) [21], [22], [23], [24]. Цікаво, що Nrf2 також регулює індукційну експресію генів цитозольних, мікросомальних і мітохондріальних форм альдегіддегідрогенази [25], які використовують NAD(P)+ як кофактор, викликаючи NAD(P)H. Дійсно, рівні NADPH і співвідношення NADPH/NADP+ нижчі в ембріональних фібробластах, виділених з Nrf2-нокаутних (Nrf2-KO) мишей, порівняно з клітинами їх побратимів дикого типу (WT), а рівні NADPH зменшуються після нокдауну Nrf2 в лінії ракових клітин з конститутивно активним Nrf2 [26]. Як і очікувалося, рівні GSH нижчі в клітинах, в яких Nrf2 був порушений; навпаки, активація Nrf2 генетичними або фармакологічними засобами призводить до підвищення GSH [27], [28], [29]. Важливо, що Nrf2 також регулює експресію генів тіоредоксину [30], [31], [32], тіоредоксинредуктази 1 [28], [29], [32], [33] та сульфіредоксину [34], які є важливими для відновлення окислених білкових тіолів.
Рисунок 2 Роль Nrf2 в метаболізмі швидко проліферуючих клітин. Nrf2 є позитивним регулятором генів, що кодують ферменти як окислювальної групи [тобто глюкозо-6-фосфатдегідрогенази (G6PD) і 6-фосфоглюконатдегідрогенази (PGD)], так і неокислювальної групи [тобто трансальдолаза 1 (TALDO1) і транскетолаза ( TKT)] пентозофосфатного шляху. G6PD і PGD генерують NADPH. Nrf2 також регулює експресію генів двох інших ферментів, що утворюють НАДФН, яблучного ферменту 1 (ME1) та ізоцитратдегідрогенази 1 (IDH1). Експресія генів фосфорибозилпірофосфат амідотрансферази (PPAT), яка каталізує вступ у шлях біосинтезу пуринів de novo, також позитивно регулюється Nrf2, так само як і експресія метилентетрагідрофолатдегідрогенази 2 (MTHFD2), яка виконує критичну роль у мітохондріях. забезпечення одновуглецевих одиниць для біосинтезу пуринів de novo. Піруваткіназа (PK) негативно регулюється Nrf2 і, як очікується, сприятиме накопиченню гліколітичних проміжних продуктів і, разом з G6PD, каналізації метаболітів через пентозофосфатний шлях і синтезу нуклеїнових кислот, амінокислот і фосфоліпідів. Nrf2 негативно регулює експресію гена АТФ-цитрат-ліази (CL), що може збільшити доступність цитрату для мітохондріальної утилізації або (через ізоцитрат) для IDH1. Червоний і синій вказують на позитивну і негативну регуляцію відповідно. Мітохондрія показана сірим кольором. Скорочення метаболітів: G-6-P, глюкозо-6-фосфат; F-6-P, фруктозо-6-фосфат; F-1,6-BP, фруктозо-1,6-біфосфат; GA-3-P, гліцеральдегід 3-фосфат; 3-PG, 3-фосфогліцерат; PEP, фосфоенолпіруват; 6-P-Gl, 6-фосфоглюконолактон; 6-PG, 6-фосфоглюконат; R-5-P, рибулозо-5-фосфат; PRPP, 5-фосфорибозил-?-1-пірофосфат; ТГФ, тетрагідрофолат; IMP, інозинмонофосфат; АМФ, аденозинмонофосфат; GMP, гуанозинмонофосфат.
Враховуючи вирішальну роль Nrf2 як головного регулятора клітинного окислювально-відновного гомеостазу, не дивно, що порівняно з клітинами WT рівні активних форм кисню (ROS) вищі в клітинах, в яких Nrf2 був порушений (Nrf2-KO). [35]. Ця різниця особливо помітна при зараженні агентами, що викликають окислювальний стрес. Крім того, клітини з дефіцитом Nrf2 набагато чутливіші до токсичності окислювачів різних типів і не можуть бути захищені індукторами Nrf2, які за тих же умов забезпечують ефективний і тривалий захист клітин WT [29], [36] , [37]. На додаток до загального клітинного окислювально-відновного гомеостазу, Nrf2 також має вирішальне значення для підтримки мітохондріального окислювально-відновного гомеостазу. Таким чином, порівняно з WT, загальний мітохондріальний пул NADH значно збільшується в Keap1-KO і різко зменшується в клітинах Nrf2-KO [35].
За допомогою візуалізації живих клітин ми нещодавно відстежували швидкість виробництва АФК у первинних гліонейрональних кокультурах і зрізах тканини мозку, виділених від мишей WT, Nrf2-KO або Keap1-нокдаун (Keap1-KD) [38]. Як і очікувалося, швидкість виробництва АФК була швидшою в клітинах і тканинах Nrf2-KO в порівнянні з їхніми аналогами WT. Проте ми зробили несподіване спостереження, що порівняно з WT клітини Keap1-KD також мають вищі показники продукції АФК, хоча величина різниці між генотипами WT та Keap1-KD була меншою, ніж між WT та Nrf2-KO. . Потім ми проаналізували рівні мРНК NOX2 і NOX4, каталітичних субодиниць двох ізоформ NADPH оксидази (NOX), які були причетні до патології мозку, і виявили, що NOX2 різко збільшується в умовах дефіциту Nrf2, тоді як NOX4 посилюється, коли Nrf2 є конститутивно активованим, хоча і в меншій мірі. У кількісному відношенні величина підвищення регуляції в клітинах і тканинах від мутантних мишей співпадає з відповідним збільшенням продукції АФК [38]. Цікаво, що Nrf2 не тільки регулює НАДФН-оксидазу, а й АФК, що виробляється НАДФН-оксидазою, може активувати Nrf2, як показано в епітеліальних клітинах легенів і кардіоміоцитах [39], [40]. Крім того, зовсім недавнє дослідження продемонструвало, що НАДФН-оксидаза-залежна активація Nrf2 є важливим ендогенним механізмом захисту від пошкодження мітохондрій і загибелі клітин у серці під час хронічного перевантаження тиском [41].
На додаток до каталітичної активності НАДФН-оксидази, мітохондріальне дихання є ще одним основним внутрішньоклітинним джерелом АФК. За допомогою мітохондрій-специфічного зонда MitoSOX ми дослідили внесок АФК мітохондріального походження в загальну продукцію АФК у первинних ізольованих гліонейрональних культурах. від мишей WT, Nrf2-KO або Keap1-KD [38]. Як і очікувалося, клітини Nrf2-KO мали вищі показники виробництва мітохондріальних АФК, ніж WT. Відповідно до висновків щодо загального виробництва АФК, показники виробництва мітохондріальних АФК у Keap1-KD також були вищими порівняно з клітинами WT. Важливо, що блокування комплексу I з ротеноном викликало різке збільшення виробництва АФК в мітохондріях як в клітинах WT, так і в Keap1-KD, але не впливало на клітини Nrf2-KO. На відміну від очікуваного збільшення мітохондріальної продукції АФК в клітинах WT після додавання пірувату (для підвищення доступності NADH, збільшення потенціалу мітохондріальної мембрани та нормалізації дихання), продукція АФК зменшилася в клітинах Nrf2-KO. Разом ці висновки свідчать про те, що за відсутності Nrf2: (i) активність комплексу I порушується, (ii) порушення активності комплексу I пов’язане з обмеженням субстратів, і (iii) порушення активності комплексу I є однією з основних причин збільшення мітохондріальної продукції АФК, можливо, через зворотний потік електронів з комплексу II.
Nrf2 впливає на потенціал мітохондріальної мембрани та дихання
Потенціал мітохондріальної мембрани (??m) є універсальним індикатором здоров’я мітохондрій і метаболічного стану клітини. У здоровій клітині ??m підтримується мітохондріальним дихальним ланцюгом. Цікаво, що стабільне ізотопне мічення амінокислотами в культуральному протеомічному дослідженні в лінії клітин MCF10A естрогенного епітелію грудної залози з негативним рецептором естрогену показало, що компонент мітохондріального ланцюга транспорту електронів NDUFA4 посилюється фармакологічною активацією (за допомогою сульфорафану, Nrroforphane). тоді як генетичне підвищення регуляції Nrf2 (за допомогою нокдауну Keap2) призводить до зниження регуляції субодиниць цитохром с оксидази COX1 і COX2I4 [1]. Дослідження протеома печінки за допомогою двовимірного гель-електрофорезу та матричної лазерної десорбції/іонізаційної мас-спектрометрії виявило, що Nrf42 регулює експресію субодиниці АТФ-синтази ? [2]. Крім того, повідомляється, що мітохондріальний білок DJ-43, який відіграє роль у підтримці активності комплексу I [1], стабілізує Nrf44 [2], [45], хоча нейропротекторні ефекти фармакологічної або генетичної активації Nrf46 не залежать від DJ-2 [1]. Однак наслідки цих спостережень для функції мітохондрій не були досліджені.
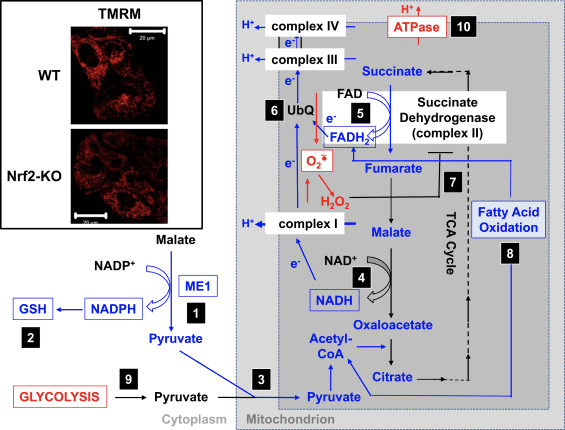
Відповідно до порушення активності комплексу I в умовах дефіциту Nrf2, базальний β?m нижчий у ембріональних фібробластах (MEFs) миші Nrf2-KO і культивованих первинних гліонейрональних клітинах порівняно з їхніми аналогами WT (рис. 3, вставка) [35]. Навпаки, базальний ??m вищий, коли Nrf2 генетично конститутивно регулюється (за допомогою нокдауну або нокауту Keap1). Ці відмінності в ??m між генотипами вказують на те, що на дихання впливає активність Nrf2. Дійсно, оцінка споживання кисню в базальному стані показала, що порівняно з WT споживання кисню нижче у МЕФ Nrf2-KO та Keap1-KO на ~50 та ~35 % відповідно.
Рисунок 3 Запропонований механізм порушення функції мітохондрій в умовах дефіциту Nrf2. (1) Зниження рівнів ME1, IDH1, G6PD і PGD призводить до зниження рівня NADPH. (2) Рівні GSH також низькі. (3) Низька активність ME1 може зменшити пул пірувату, що надходить у мітохондрії. (4) Генерація NADH відбувається повільніше, що призводить до порушення активності комплексу I і збільшення продукції АФК мітохондріями. (5) Відновлення FAD до FADH2 в мітохондріальних білках також зменшується, знижуючи потік електронів від FADH2 до UbQ і в комплекс III. (6) Повільніше утворення UbQH2 може знизити активність ферменту сукцинатдегідрогенази. (7) Підвищені рівні АФК можуть додатково інгібувати активність комплексу II. (8) Нижча ефективність окислення жирних кислот сприяє зниженню доступності субстрату для мітохондріального дихання. (9) Гліколіз посилюється як компенсаторний механізм для зниження виробництва АТФ при окисному фосфорилюванні. (10) АТФ-синтаза діє у зворотному порядку, щоб підтримувати ??m. Червоний і синій вказують на підвищення та зниження регуляції відповідно. Коробки означають наявність експериментальних доказів. На вставці показані зображення мітохондрій кортикальних астроцитів WT і Nrf2-KO, візуалізовані за допомогою потенціометричного флуоресцентного зонда метилового ефіру тетраметилродаміну (TMRM; 25 нМ). Масштабна смуга, 20 м.
Ці відмінності в ??m і диханні між генотипами відображаються швидкістю використання субстратів для мітохондріального дихання. Застосування субстратів для циклу трикарбонової кислоти (TCA) (малат/піруват, які, у свою чергу, збільшують продукцію субстрату комплексу I NADH) або метилсукцинату, субстрату для комплексу II, спричиняє ступінчасте збільшення ??m в обох WT і нейрони Keap1-KD, але швидкість збільшення вища в клітинах Keap1-KD. Що ще важливіше, форми відповіді на ці субстрати циклу TCA відрізняються між двома генотипами, завдяки чому швидке підвищення ??m в клітинах Keap1-KD після додавання субстрату супроводжується швидким падінням, а не плато, що свідчить про незвичайну швидке споживання субстрату. Ці результати тісно узгоджуються з набагато нижчими (на 50%) рівнями малату, пірувату і сукцинату, які спостерігалися після 70-годинного імпульсу [U-1C13]глюкози в Keap6-KO в порівнянні з WT MEF. клітини [1]. У нейронах Nrf24-KO тільки піруват здатний збільшувати β?m, тоді як малат і метилсукцинат викликають помірну деполяризацію. Вплив Nrf2 на продукцію мітохондріального субстрату, здається, є основним механізмом, за допомогою якого Nrf2 впливає на функцію мітохондрій. Редокс-індекс мітохондріального NADH (баланс між споживанням NADH комплексом I та продукцією NADPH у циклі TCA) значно нижчий у клітинах Nrf2-KO порівняно з їхніми аналогами WT, а також швидкість регенерації пулів NADH і FADH2 після інгібування комплексу IV (за допомогою NaCN) діють повільніше в мутантних клітинах.
У мітохондріях, ізольованих з мозку та печінки мишей, додавання субстратів для комплексу I або для комплексу II сильніше збільшує швидкість споживання кисню, коли Nrf2 активується, і менш ефективно, коли Nrf2 порушується [35]. Таким чином, малат викликає вищу швидкість споживання кисню в Keap1-KD порівняно з WT, але його ефект слабший в мітохондріях Nrf2-KO. Аналогічно, у присутності ротенону (коли комплекс I інгібується), сукцинат активує споживання кисню більшою мірою в Keap1-KD порівняно з WT, тоді як відповідь у мітохондріях Nrf2-KO зменшується. Крім того, первинні нейрональні культури Nrf2-KO та миші є більш чутливими до токсичності інгібіторів комплексу II 3-нітропропіонової кислоти та малонату, тоді як внутрішньостриарна трансплантація астроцитів, що надекспресують Nrf2, є захисною [48], [49]. Аналогічно, миші Nrf2-KO більш чутливі до нейротоксичності, спричиненої інгібітором комплексу I 2-метил-1-фенілпіридинію в іоні 4-метил-1-феніл-4, тоді як генетична або фармакологічна активація Nrf1,2,3,6 має захисний ефект. 49-тетрагідропіридинова модель хвороби Паркінсона на тваринах [50], [51], [52], [53], [54], [55], [56], [57], [58], [59], [60], [61], [XNUMX].
Коефіцієнт респіраторного контролю (RCR), відношення стану 3 (стимульований ADP) до стану 4 (відсутня ADP), зменшується за відсутності Nrf2, але RCR подібний між мітохондріями Keap1-KD і WT [35 ]. Оскільки RCR є показником ступеня зв’язку активності мітохондріального дихального ланцюга з окисним фосфорилюванням, цей висновок вказує на те, що вища швидкість дихання в мітохондріях Keap1-KD не пов’язана з роз’єднанням окисного фосфорилювання. Це також свідчить про те, що окисне фосфорилювання є більш ефективним, коли Nrf2 активований. Вища швидкість дихання в мітохондріях Keap1-KD узгоджується з вищими рівнями виробництва мітохондріальних АФК [38], оскільки більша швидкість дихання може призвести до збільшення витоку електронів. Однак в умовах окисного стресу підвищеній продукції АФК протидіє Nrf2-залежна транскрипційна регуляція роз’єднувального білка 3 (UCP3), яка підвищує протонну провідність внутрішньої мембрани мітохондрій і, отже, зменшує продукцію супероксиду [62]. Зовсім недавно було показано, що продукт перекисного окислення ліпідів 4-гідрокси-2-ноненал опосередковує Nrf2-залежну регуляцію UCP3 в кардіоміоцитах; це може бути особливо важливим для захисту в умовах окисного стресу, наприклад під час реперфузії ішемії [63].
Nrf2 впливає на ефективність окисного фосфорилювання та синтез АТФ
Відповідно до впливу Nrf2 на дихання, у мітохондріях мозку та печінки, дефіцит Nrf2 призводить до зниження ефективності окисного фосфорилювання (за оцінкою відношення АДФ до кисню, який витрачається на синтез АТФ), тоді як активація Nrf2 (Keap1 -KD) має протилежний ефект [35]. Порівняно з WT, рівні АТФ значно вищі в клітинах з конститутивною регуляцією Nrf2 і нижчі, коли Nrf2 знищений [64] або порушений [35]. Крім того, використання інгібіторів окисного фосфорилювання (олігоміцин) або гліколізу (йодооцтова кислота) показало, що Nrf2 змінює спосіб, за допомогою якого клітини виробляють АТФ. Таким чином, в нейронах WT олігоміцин викликає повне падіння АТФ і йодооцтова кислота не має подальшого впливу. Примітно, що в клітинах Nrf2-KO олігоміцин підвищує рівень АТФ, який потім повільно, але повністю виснажується йодооцтовою кислотою, що вказує на те, що за відсутності Nrf2 основним джерелом виробництва АТФ є гліколіз, а не окисне фосфорилювання. Цікаво, що незважаючи на підвищену ефективність окисного фосфорилювання в клітинах Keap1-KD, додавання олігоміцину призводить до ~80% зниження рівня АТФ, а йодооцтова кислота викликає подальше зниження на ~20%. Таким чином, або дефіцит Nrf2, або його конститутивна активація зменшує внесок окисного фосфорилювання і збільшує внесок гліколізу в синтез АТФ. Цей ефект особливо виражений, коли Nrf2 відсутній і узгоджується із залежністю ??m від присутності глюкози в середовищі [35] і підвищеними рівнями гліколітичних проміжних продуктів (G-6-P, F-6-P , дигідроксиацетонфосфат, піруват і лактат) після нокдауну Nrf2 [24].
Підвищення рівня АТФ після інгібування F1F0-АТФази олігоміцином вказує на те, що за відсутності Nrf2 F1F0-АТФаза функціонує як АТФаза, а не АТФ-синтаза, тобто діє у зворотному порядку. Така зміна активності, швидше за все, відображає необхідність перекачування протонів через внутрішню мітохондріальну мембрану в спробі зберегти ??m, що має вирішальне значення для функціональної цілісності цієї органели. Про зміну функції F1F0-АТФази також свідчить спостережувана деполяризація мітохондрій при введенні олігоміцину в клітини Nrf2-KO, що різко контрастує з гіперполяризацією, що виникає в їхніх WT або Keap1-дефіцитних аналогах [35]. Загалом, схоже, що в умовах дефіциту Nrf2 АТФ виробляється в основному в результаті гліколізу, і цей АТФ потім частково використовується F1F0-АТФазою для підтримки ??m.
Вплив дефіциту Nrf2 на ??m особливо виражений, коли клітини інкубують у середовищі без глюкози, а ??m є на ~50% нижчим у Nrf2-KO порівняно з клітинами WT [35]. В умовах нестачі глюкози окислення мітохондріальних жирних кислот (FAO) є основним постачальником субстратів для дихання та окисного фосфорилювання, що свідчить про те, що Nrf2 може впливати на FAO. Справді, ефективність FAO як для довголанцюгової (C16:0) насиченої жирної кислоти пальмітинової кислоти, так і для коротколанцюгової (C6:0) гексанової кислоти вища в Keap1-KO MEFs та ізольованих мітохондріях серця і печінки, ніж у їх WT аналогів, тоді як він нижчий у клітинах Nrf2-KO та мітохондріях [65]. Ці ефекти також мають велике значення для людей: дійсно, метаболічні зміни, що вказують на кращу інтеграцію FAO з активністю циклу TCA, як повідомлялося, відбуваються в дослідженнях людського втручання з дієтами, багатими на глюкорафанін, попередник класичного активатора Nrf2 сульфорафану [ 66].
Під час першого етапу мітохондріальної FAO про-R водень ?-вуглецю виходить у вигляді гідриду, який відновлює кофактор FAD до FADH2, який, у свою чергу, передає електрони на убіхінон (UbQ) у дихальному ланцюгу, що в кінцевому підсумку сприяє виробленню АТФ. . У той час як стимуляція FAO пальмітоілкарнітином за відсутності глюкози викликає очікуване підвищення рівня АТФ в клітинах WT і Keap1-KO, при цьому зростання АТФ відбувається швидше в клітинах Keap1-KO, ідентична обробка не викликає змін АТФ в Nrf2-KO. MEFs [65]. Цей експеримент демонструє, що за відсутності Nrf2 ФАО пригнічується, і, крім того, це передбачає придушення ФАО як одну з причин зниження рівня АТФ в умовах дефіциту Nrf2 [35], [64].
Примітно, що людські 293 Т-клітини, в яких Nrf2 був заглушений, мають нижчу експресію CPT1 і CPT2[67], двох ізоформ карнітинпальмітоілтрансферази (CPT), ферменту, що обмежує швидкість в мітохондріальних FAO. Відповідно, рівні мРНК Cpt1 нижчі в печінці Nrf2-KO порівняно з мишами WT [68]. CPT каталізує перенесення ацильної групи довголанцюгового жирного ацил-КоА від коензиму А до L-карнітину і таким чином дозволяє імпортувати ацилкарнітин з цитоплазми в мітохондрії. Хоча це не було досліджено на сьогоднішній день, можливо, що на додаток до транскрипційного впливу на експресію CPT1, Nrf2 також може впливати на функцію цього ферменту, контролюючи рівні його основного алостеричного інгібітора, малоніл-КоА. Це пояснюється тим, що за механізмом, який наразі неясний, Nrf2 негативно регулює експресію стеароіл-КоА-десатурази (SCD) [69] і цитрат-ліази (CL) [69], [70]. Цікаво, що нокаут або інгібування SCD призводить до посилення фосфорилювання та активації AMP-активованої протеїнкінази (AMPK) [71], [72], [73], і можна припустити, що за відсутності Nrf2 рівні SCD підвищиться, у свою чергу, знизить активність AMPK. Це може бути додатково посилено зниженими рівнями білка AMPK, які спостерігалися в печінці мишей Nrf2-KO [68], що тісно узгоджується з підвищеними рівнями AMPK, про які повідомлялося в печінці Keap1-KD. миші [74]. Одним із наслідків зниженої активності AMPK є полегшення його інгібуючого фосфорилювання (на Ser79) ацетил-КоА карбоксилази (ACC) [75], яке може бути додатково підвищене транскрипції за відсутності Nrf2, оскільки воно зменшується активацією Nrf2 [70]. ]. Висока активність ACC у поєднанні з підвищеною експресією CL, що збільшує вироблення ацетил-КоА, субстрату для ACC, може в кінцевому підсумку підвищити рівень продукту ACC, малоніл-КоА. Високі рівні малоніл-КоА інгібують CPT, тим самим зменшуючи транспорт жирних кислот у мітохондрії. Нарешті, Nrf2 позитивно регулює експресію CD36 [76], транслокази, яка імпортує жирні кислоти через плазматичні та мітохондріальні мембрани. Таким чином, одним з механізмів, за допомогою якого Nrf2 може впливати на ефективність мітохондріальної FAO, є регулювання імпорту довголанцюгових жирних кислот в мітохондрії.
На додаток до прямої регуляції транскрипції, Nrf2 також може змінювати ефективність мітохондріальної FAO через його вплив на клітинний окислювально-відновний метаболізм. Це може бути особливо актуальним, коли активність Nrf2 низька або відсутня, умови, які зміщують клітинний окислювально-відновний статус у бік окисленого стану. Дійсно, кілька ферментів ФАО було ідентифіковано як чутливі до окислювально-відновних змін. Одним з таких ферментів є дуже довголанцюгова ацил-КоА-дегідрогеназа (VLCAD), яка сприяє більш ніж 80% активності дегідрування пальмітоїл-КоА в тканинах людини [77]. Цікаво, що Hurd et al. [78] показали, що VLCAD містить залишки цистеїну, які значно змінюють свій окислювально-відновний стан під час впливу H2O2 на ізольовані мітохондрії серця щура. Крім того, S-нітрозилування VLCAD печінки мишей на Cys238 покращує каталітичну ефективність ферменту [79], і цілком ймовірно, що окислення того ж цистеїну може мати протилежний ефект, зрештою знижуючи ефективність мітохондріальної FAO. Таким чином, можливо, що, хоча рівні експресії VLCAD істотно не відрізняються в МЕФ WT, Nrf2-KO або Keap1-KO [65], активність ферменту VLCAD може бути нижчою за відсутності Nrf2 через більш високі рівні. ROS.
На основі всіх цих висновків можна припустити, що (рис. 3): за відсутності Nrf2 рівні NADPH нижчі через зниження експресії ME1, IDH1, G6PD та PGD. Рівні відновленого глутатіону також нижчі через зниження експресії ферментів, які беруть участь у його біосинтезі та регенерації, та нижчих рівнів NADPH, які необхідні для перетворення окисленої у відновлену форму глутатіону. Низька експресія ME1 зменшить пул пірувату, що надходить у мітохондрії, при цьому гліколіз стане основним джерелом пірувату. Генерація NADH відбувається повільніше, що призводить до порушення активності комплексу I і збільшення продукції АФК мітохондріями. Відновлення FAD до FADH2 також повільніше, принаймні частково через менш ефективне окислення жирних кислот, що порушує потік електронів від FADH2 до UbQ і в комплекс III. Оскільки UbQH2 є активатором сукцинатдегідрогенази [80], уповільнення його утворення може знизити активність ферменту сукцинатдегідрогенази. Підвищені рівні супероксиду та перекису водню можуть додатково інгібувати активність комплексу II [81]. Нижча ефективність окислення жирних кислот сприяє зниженню доступності субстрату для мітохондріального дихання та виробництва АТФ при окисному фосфорилюванні. Як компенсаторний механізм посилюється гліколіз. АТФ-синтаза функціонує у зворотному порядку, як АТФаза, намагаючись підтримувати ??m.
Nrf2 і мітохондріальний біогенез
Повідомлялося, що порівняно з WT печінка мишей Nrf2-KO має нижчий вміст мітохондрій (що визначається співвідношенням мітохондріальної та ядерної ДНК); це ще більше зменшується 24-годинним голодуванням у мишей WT і Nrf2-KO; навпаки, хоча й не відрізняється від WT за нормальних умов годування, вміст мітохондрій у мишей з високою активністю Nrf2 не впливає на голодування [82]. Цікаво, що добавка активатора Nrf2 (R)-?-ліпоєвої кислоти [83], [84], [85] сприяє мітохондріальному біогенезу в адипоцитах 3T3-L1 [86]. Два класи ядерних регуляторів транскрипції відіграють вирішальну роль у мітохондріальному біогенезі. До першого класу належать фактори транскрипції, такі як ядерні респіраторні фактори11 і 2, які контролюють експресію генів, що кодують субодиниці п’яти дихальних комплексів, мітохондріальних трансляційних компонентів і біосинтетичних ферментів гему, які локалізовані в мітохондріальному матриксі [88]. Piantadosi та ін. [89] показали, що Nrf2-залежна транскрипційна регуляція ядерного респіраторного фактора 1 сприяє мітохондріальному біогенезу та захищає від цитотоксичності кардіотоксичного антрациклінового хіміотерапевтичного засобу доксорубіцину. На противагу цьому, Zhang et al. [82] повідомили, що генетична активація Nrf2 не впливає на експресію базальної мРНК ядерного респіраторного фактора 1 у печінці мишей.
Другим класом ядерних регуляторів транскрипції з критичними функціями в мітохондріальному біогенезі є коактиватори транскрипції, такі як рецептор, активований проліфератором пероксисом ? коактиватори (PGC)1? і 1?, які взаємодіють з факторами транскрипції, механізмами базальної транскрипції та РНК-сплайсингу, а також ферментами, що модифікують гістон [88], [90], [91]. На експресію сімейства коактиваторів PGC1 впливають численні сигнали навколишнього середовища. Обробка фібробластів людини активатором Nrf2 сульфорафаном викликає збільшення маси мітохондрій та індукцію PGC1? і PGC1? [92], хоча потенційна залежність від Nrf2 у цьому дослідженні не досліджувалася. Однак миші з цукровим діабетом, у яких Nrf2 або активований гіпоморфним нокдауном гена Keap1 (db/db:Keap1flox/?:Nrf2+/+), або порушений (db/db:Keap1flox/?:Nrf2?/?), мають нижчий печінковий PGC1? рівні експресії, ніж у контрольних тварин (db/db:Keap1flox/+:Nrf2+/+) [93]. Немає відмінностей у рівнях мРНК для PGC1? спостерігаються в печінці недіабетичних мишей, які мають або WT, або Nrf2-KO, тоді як ці рівні нижчі у тварин з надмірною експресією Nrf2 (Keap1-KD і специфічний для печінки Keap1-KO) [82]. Примітно, що 24-годинне голодування підвищує рівень PGC1? мРНК у печінці мишей усіх генотипів, але збільшення значно більше в печінці Nrf2-KO порівняно з мишами з надекспресією WT або Nrf2. Порівняно з WT, миші Nrf2-KO, у яких зазнала септична інфекція або гостре ураження легень через інфекцію, демонструють ослаблену транскрипційну регуляцію ядерного респіраторного фактора 1 і PGC1? [94], [95]. Разом ці спостереження свідчать про те, що роль Nrf2 у підтримці рівнів як ядерного респіраторного фактора 1, так і PGC1? є складним і стає найбільш помітним в умовах стресу.
Крім експресії генів, що кодують мітохондріальні білки, біогенез мітохондрій вимагає синтезу нуклеотидів. Генетична активація Nrf2 посилює біосинтез пуринів шляхом активізації пентозофосфатного шляху та метаболізму фолатів і глутаміну, особливо в клітинах, що швидко проліферують (рис. 2) [24]. Аналіз транскриптому мутантної дрозофіли з дефіцитом мітохондріальної серин/треонін протеїнкінази PTEN-індукованої передбачуваної кінази 1 (PINK1) показав, що мітохондріальна дисфункція призводить до активізації транскрипції генів, що впливають на метаболізм нуклеотидів, що свідчить про посилення біонуклеотидів [96]. являє собою механізм захисту від нейротоксичних наслідків дефіциту PINK1. Nrf2 регулює експресію фосфорибозилпірофосфат амідотрансферази (PPAT), яка каталізує входження в шлях біосинтезу пуринових нуклеотидів de novo, і мітохондріальної метилентетрагідрофолатдегідрогенази 2 (MTHFD2) (рис. 2). Останній є біфункціональним ферментом з активністю дегідрогенази та циклогідролази, який має вирішальне значення для забезпечення як гліцину, так і формиату як джерела одновуглецевих одиниць для біосинтезу пуринів у швидкозростаючих клітинах [97]. Тому імовірно, що активація Nrf2 може бути захисною і може повернути мітохондріальну дисфункцію при дефіциті PINK1. Дійсно, фармакологічна активація Nrf2 сульфорафаном або тритерпеноїдом RTA-408 відновлює ??m і захищає PINK1-дефіцитні клітини від токсичності дофаміну [98]. Хоча основні механізми здаються складними, разом ці дані вказують на те, що активність Nrf2 може впливати на біогенез мітохондрій, впливаючи на рівні експресії критичних транскрипційних факторів і коактиваторів, а також шляхом посилення біосинтезу нуклеотидів.
Nrf2 і цілісність мітохондрій
Хоча прямі докази не завжди доступні, є серйозні ознаки того, що Nrf2 важливий для цілісності мітохондрій, особливо в умовах окисного стресу. Мітохондрії, виділені з мозку та печінки щурів, яким ввели одноразову дозу сульфорафану-активатора Nrf2, стійкі до відкриття пори переходу проникності мітохондрій (mPTP), викликаного окислювачем трет-бутилгідропероксидом [99], [100]. Нещодавно було виявлено, що mPTP, комплекс, який дозволяє внутрішній мембрані мітохондрій стати проникною для молекул з масою до 1500 Да, утворюється з димерів F0F1-АТФ-синтази [101]. Опосередкована сульфорафаном резистентність до відкриття mPTP корелює з підвищеним антиоксидантним захистом, а рівні мітохондріального GSH, глутатіонпероксидази 1, яблучного ферменту 3 і тіоредоксину 2 підвищуються у фракціях мітохондрій, виділених у тварин, які отримували сульфорафан [100].
Пошкодження мітохондріальних білків і порушення дихання, викликані продуктом електрофільного перекисного окислення ліпідів 4-гідрокси-2-ноненаль, ослаблені в мітохондріях, виділених з кори головного мозку мишей, які отримували сульфорафан [102]. В епітеліальних клітинах нирок щурів і в нирках сульфорафан захищає від токсичності, спричиненої цисплатином і гентаміцином, і втрати β?m[103], [104]. Захист від панелі окислювачів (супероксид, перекис водню, пероксинітрит) і електрофілів (4-гідрокси-2-ноненал і акролеїн) і підвищення мітохондріального антиоксидантного захисту також спостерігали при обробці гладком’язових клітин аорти щурів сульфорафаном [105]. ]. У моделі гострого ураження нирок, індукованого контрастом, нещодавно було показано, що ішемічне прекондиціонування кінцівок має захисні ефекти, включаючи інгібування відкриття mPTP і набряку мітохондрій шляхом активації Nrf2 внаслідок інгібування GSK3? [106].
Мітофагія, процес, за допомогою якого дисфункціональні мітохондрії вибірково поглинаються аутофагосомами і доставляються в лізосоми для деградації та перероблення клітиною, має важливе значення для мітохондріального гомеостазу [107], [108]. Хоча причинний зв’язок між Nrf2 та мітофагією не встановлено, є докази того, що фактор транскрипції може бути важливим у контролі якості мітохондрій, відіграючи певну роль у мітофагії. Це може бути особливо помітним в умовах окисного стресу. Так, у моделі сепсису підвищення рівнів маркера аутофагосоми MAP1 легкого ланцюга 3-II (LC3-II) і білка вантажу p62 через 24 години після інфекції пригнічується у Nrf2-KO порівняно з мишами WT [109] . Нещодавно було відкрито маломолекулярний індуктор мітофагії (так званий p62-опосередкований індуктор мітофагії, PMI); ця сполука 1,4-дифеніл-1,2,3-триазолу спочатку була розроблена як активатор Nrf2, який порушує взаємодію фактора транскрипції з Keap1 [110]. Подібно до клітин, в яких Nrf2 генетично регулюється (Keap1-KD або Keap1-KO), клітини, що піддаються впливу PMI, мають більш високий рівень спокою. Важливо, що збільшення мітохондріальної локалізації LC3, яке спостерігається після обробки PMI клітин WT, не відбувається в клітинах Nrf2-KO, що свідчить про залучення Nrf2.
Нарешті, ультраструктурний аналіз зрізів печінки виявив наявність набряклих мітохондрій із зменшеною кристою та зруйнованими мембранами в гепатоцитах Nrf2-KO, але не WT, мишей, яких годували дієтою з високим вмістом жирів протягом 24 тижнів; зокрема, ця печінка демонструє чіткі ознаки окисного стресу та запалення [68]. Можна зробити висновок, що Nrf2 відіграє вирішальну роль у підтримці цілісності мітохондрій в умовах окисного та запального стресу.
Сульфорафан та його вплив на рак, смертність, старіння, мозок і поведінку, хвороби серця тощо
Ізотіоціанати є одними з найважливіших рослинних сполук, які ви можете отримати у своєму раціоні. У цьому відео я роблю для них найповнішу справу, яку коли-небудь робили. Короткий період уваги? Перейдіть до улюбленої теми, натиснувши один із моментів часу нижче. Повна хронологія нижче.
Ключові розділи:
00:01:14 – Рак і смертність
00:19:04 – Старіння
00:26:30 – Мозок і поведінка
00:38:06 – Підсумок
00:40:27 – Доза
Повний графік:
00:00:34 – Представлення сульфорафану, головна тема відео.
00:01:14 – Споживання овочів хрестоцвітних і зниження смертності від усіх причин.
00:02:12 – Ризик раку передміхурової залози.
00:02:23 – Ризик раку сечового міхура.
00:02:34 – Ризик раку легенів у курців.
00:02:48 – Ризик раку молочної залози.
00:03:13 – Гіпотетична: що робити, якщо у вас уже рак? (інтервенційний)
00:03:35 – Імовірний механізм, який керує асоціативними даними раку та смертності.
00:04:38 – Сульфорафан і рак.
00:05:32 – Докази на тваринах, що показують сильний вплив екстракту паростків брокколі на розвиток пухлин сечового міхура у щурів.
00:06:06 – Вплив прямого прийому сульфорафану у пацієнтів з раком передміхурової залози.
00:07:09 – Біоакумуляція метаболітів ізотіоціаната у фактичній тканині молочної залози.
00:08:32 – Пригнічення стовбурових клітин раку молочної залози.
00:08:53 – Урок історії: ще в Стародавньому Римі стверджували, що капустяні гриби мають оздоровчі властивості.
00:09:16 – Здатність сульфорафану посилювати виведення канцерогену (бензолу, акролеїну).
00:09:51 – NRF2 як генетичний перемикач через елементи антиоксидантної реакції.
00:10:10 – Як активація NRF2 посилює виведення канцерогену через глутатіон-S-кон'югати.
00:10:34 – Брюссельська капуста підвищує глутатіон-S-трансферазу і зменшує пошкодження ДНК.
00:11:20 – Напій з проростків брокколі збільшує виведення бензолу на 61%.
00:15:45 – Споживання хрестоцвітних овочів і смертність від серцево-судинних захворювань.
00:16:55 – порошок паростків брокколі покращує рівень ліпідів у крові та загальний ризик серцевих захворювань у діабетиків 2 типу.
00:19:04 – Початок секції старіння.
00:19:21 – Дієта, збагачена сульфорафаном, збільшує тривалість життя жуків від 15 до 30% (за певних умов).
00:20:34 – Важливість слабкого запалення для довголіття.
00:22:05 – Овочі хрестоцвітних і порошок паростків брокколі, здається, зменшують широкий спектр запальних маркерів у людей.
00:23:40 – Підсумок у середині відео: розділи про рак, старіння
00:24:14 – Дослідження на мишах показують, що сульфорафан може покращити адаптивну імунну функцію в літньому віці.
00:25:18 – Сульфорафан покращив ріст волосся у мишачої моделі облисіння. Зображення на 00:26:10.
00:26:30 – Початок розділу «Мозок і поведінка».
00:27:18 – Вплив екстракту паростків брокколі на аутизм.
00:27:48 – Вплив глюкорафаніну на шизофренію.
00:28:17 – Початок обговорення депресії (правдоподібний механізм та дослідження).
00:31:21 – Дослідження на мишах з використанням 10 різних моделей депресії, викликаної стресом, показало, що сульфорафан так само ефективний, як і флуоксетин (прозак).
00:32:00 – Дослідження показує, що пряме вживання глюкорафаніну мишами так само ефективне для запобігання депресії через модель стресу соціальної поразки.
00:33:01 – Початок відділу нейродегенерації.
00:33:30 – Сульфорафан і хвороба Альцгеймера.
00:33:44 – Сульфорафан і хвороба Паркінсона.
00:33:51 – Сульфорафан і хвороба Хантінгтона.
00:34:13 – Сульфорафан збільшує кількість білків теплового шоку.
00:34:43 – Початок секції черепно-мозкової травми.
00:35:01 – Сульфорафан, введений відразу після ЧМТ, покращує пам’ять (дослідження на мишах).
00:35:55 – Сульфорафан і нейрональна пластичність.
00:36:32 – Сульфорафан покращує навчання на моделі діабету ІІ типу у мишей.
00:37:19 – Сульфорафанова і м’язова дистрофія Дюшенна.
00:37:44 – Інгібування міостатину в клітинах-супутниках м’язів (in vitro).
00:38:06 – Пізнє відео: смертність і рак, пошкодження ДНК, окислювальний стрес і запалення, виділення бензолу, серцево-судинні захворювання, діабет ІІ типу, вплив на мозок (депресія, аутизм, шизофренія, нейродегенерація), шлях NRF2.
00:40:27 – Думки щодо визначення дози паростків брокколі або сульфорафану.
00:41:01 – Анекдоти про проростання в домашніх умовах.
00:43:14 – Про температуру приготування та активність сульфорафану.
00:43:45 – Перетворення сульфорафану з глюкорафаніну кишковими бактеріями.
00:44:24 – Добавки працюють краще в поєднанні з активною мирозиназою з овочів.
00:44:56 – Техніка приготування та овочі хрестоцвітних.
00:46:06 – Ізотіоціанати як зоб.
Nrf2 є фактором транскрипції, який відіграє важливу роль у системі клітинного антиоксидантного захисту організму людини. Елемент, що реагує на антиоксиданти, або ARE, є регуляторним механізмом генів. Багато досліджень продемонстрували, що Nrf2 або фактор 2, пов’язаний з NF-E2, регулює широкий спектр генів, керованих ARE, у кількох типах клітин. Було також виявлено, що Nrf2 відіграє важливу роль у захисті клітин та антиканцерогенності, що демонструє, що Nrf2 може бути ефективним засобом лікування нейродегенеративних захворювань та раку, які, як вважають, спричинені окислювальним стресом. Д-р Алекс Хіменес, округ Колумбія, CCST Insight
Заключні зауваження
Хоча багато питань все ще залишаються відкритими, наявні експериментальні дані чітко вказують на те, що Nrf2 є важливим гравцем у підтримці мітохондріального гомеостазу та структурної цілісності. Ця роль стає особливо важливою в умовах окисного, електрофільного та запального стресу, коли здатність посилювати опосередковані Nrf2 цитопротекторні реакції впливає на загальне здоров’я та виживання клітини та організму. Роль Nrf2 у функції мітохондрій являє собою ще один шар широких цитопротекторних механізмів, оркестрованих цим фактором транскрипції. Оскільки багато патологічних станів людини мають окислювальний стрес, запалення та мітохондріальну дисфункцію як важливі компоненти їх патогенезу, фармакологічна активація Nrf2 є перспективною для профілактики та лікування захворювань. Всебічне розуміння точних механізмів, за допомогою яких Nrf2 впливає на функцію мітохондрій, є важливим для раціонального планування майбутніх клінічних досліджень і може запропонувати нові біомаркери для моніторингу терапевтичної ефективності.
Метою статті вище було обговорити, а також продемонструвати роль Nrf2 у функції мітохондрій. Nrf2, або ядерний фактор, пов'язаний з еритроїдом 2, є новим регулятором стійкості клітин до окисників, які можуть сприяти окислювальному стресу, впливаючи на клітинну функцію і призводячи до розвитку токсичності, хронічних захворювань і навіть раку. Хоча виробництво окислювачів в організмі людини може служити для різних цілей, включаючи поділ клітин, запалення, імунну функцію, аутофагію та реакцію на стрес, важливо контролювати їх перевиробництво, щоб запобігти проблемам зі здоров’ям. Обсяг нашої інформації обмежено хіропрактикою та проблемами здоров’я хребта. Щоб обговорити тему, зверніться до доктора Хіменеса або зв’яжіться з нами за адресою�915-850-0900.
Додаткова тема для обговорення: «Гострий біль у спині».
Біль у спині� є однією з найпоширеніших причин інвалідності та пропущених робочих днів у всьому світі. Біль у спині є другою за поширеністю причиною відвідувань лікаря, переважаючи лише інфекції верхніх дихальних шляхів. Приблизно 80 відсотків населення відчувають біль у спині хоча б раз у житті. Хребет – це складна структура, що складається з кісток, суглобів, зв’язок і м’язів, серед інших м’яких тканин. Через це травми та/або загострення стану, такі як�грижі диски, може зрештою призвести до симптомів болю в спині. Спортивні травми або травми в автомобільній катастрофі часто є найчастішою причиною болю в спині, однак іноді найпростіші рухи можуть мати хворобливі наслідки. На щастя, альтернативні варіанти лікування, такі як хіропрактика, можуть допомогти полегшити біль у спині за допомогою корекції хребта та ручних маніпуляцій, що в кінцевому підсумку покращує полегшення болю. �
Nrf2 підтримує активацію групи антиоксидантних і детоксикаційних ферментів і генів, які захищають організм людини від наслідків проблем зі здоров’ям, пов’язаних із підвищеним рівнем окисного стресу, наприклад, хвороба Альцгеймера. Було продемонстровано, що різноманітні природні речовини активують шлях Nrf2, що може допомогти впоратися з симптомами нейродегенеративних захворювань. Метою статті нижче є обговорення ключової ролі Nrf2, спричиненої хронічним запаленням.
абстрактний
Запалення є найбільш поширеною ознакою багатьох хронічних захворювань і ускладнень, відіграючи важливу роль у канцерогенезі. Кілька досліджень продемонстрували, що Nrf2 сприяє протизапальному процесу, організовуючи рекрутинг запальних клітин і регулюючи експресію генів за допомогою елемента антиоксидантної відповіді (ARE). Keap1 (Kelch-подібний ECH-асоційований білок)/Nrf2 (NF-E2 p45-пов'язаний фактор 2)/ARE сигнальний шлях в основному регулює експресію протизапальних генів і пригнічує прогресування запалення. Тому визначення нових Nrf2-залежних протизапальних фітохімічних речовин стало ключовим моментом у відкритті ліків. У цьому огляді ми обговорюємо членів сигнального шляху Keap1/Nrf2/ARE та його гени, що знаходяться нижче, вплив цього шляху на тваринні моделі запальних захворювань та перехресні зв’язки зі шляхом NF-?B. Крім того, ми також обговорюємо регуляцію інфламасоми NLRP3 за допомогою Nrf2. Крім цього, ми підсумовуємо поточний сценарій розвитку протизапальних фітохімічних речовин та інших, які опосередковують сигнальний шлях Nrf2/ARE.
Запалення – це складний процес, який виникає, коли тканини інфікуються або пошкоджуються шкідливими подразниками, такими як патогени, пошкодження або подразники. У цій захисній реакції беруть участь імунні клітини, кровоносні судини та молекулярні медіатори [1]. Запалення також є патологічним явищем, пов’язаним з різноманітними хворобливими станами, спричиненими переважно фізичними, хімічними, біологічними та психологічними факторами. Метою запалення є обмеження та усунення причин пошкодження клітин, очищення та/або поглинання некротичних клітин і тканин та ініціювання відновлення тканин. Розрізняють дві форми запалення: гостру і хронічну. Гостре запалення самообмежується і є корисним для господаря, але тривале хронічне запалення є загальною ознакою багатьох хронічних захворювань і ускладнень. Пряма інфільтрація багатьма мононуклеарними імунними клітинами, такими як моноцити, макрофаги, лімфоцити та плазматичні клітини, а також вироблення запальних цитокінів призводять до хронічного запалення. Визнано, що хронічне запалення відіграє вирішальну роль у канцерогенезі [2]. Загалом, як про-, так і протизапальні сигнальні шляхи взаємодіють у нормальному запальному процесі.
При патологічному запальному процесі спочатку активізуються тучні клітини, моноцити, макрофаги, лімфоцити та інші імунні клітини. Потім клітини залучаються до місця пошкодження, що призводить до утворення активних форм кисню (АФК), які пошкоджують макромолекули, включаючи ДНК. У той же час ці запальні клітини також виробляють велику кількість медіаторів запалення, таких як цитокіни, хемокіни та простагландини. Ці медіатори додатково залучають макрофаги до локалізованих місць запалення і безпосередньо активують численні каскади передачі сигналу та транскрипційні фактори, пов’язані із запаленням. Сигнальні шляхи NF-?B (ядерний фактор каппа B), MAPK (мітоген-активована протеїнкіназа) і JAK (янус-кіназа)-STAT (перетворювачі сигналів і активатори транскрипції) беруть участь у розвитку класичного шляху запалення. [3], [4], [5]. Попередні дослідження показали, що фактор транскрипції Nrf2 (NF-E2 p45-пов’язаний фактор 2) регулює експресію ферментів фази II детоксикації, включаючи NADPH, NAD(P)H хіноноксидоредуктазу 1, глутатіонпероксидазу, феритин, гемоксигеназу-1 (HO -1), а також антиоксидантні гени, які захищають клітини від різних ушкоджень за допомогою своїх протизапальних ефектів, таким чином впливаючи на перебіг захворювання [6], [7], [8].
Враховуючи ці чудові висновки, останніми роками великий інтерес викликала розробка цільових терапевтичних препаратів для лікування запальних захворювань через сигнальні шляхи. У цьому огляді ми підсумовуємо дослідження сигнального шляху Keap1 (білок, подібний до ECH)/Nrf2 (фактор 2, пов’язаний з NF-E45 p2)/ARE (елемент антиоксидантної реакції) при запаленні.
Структура та регулювання Nrf2
Регулювання Nrf1, залежне від Keap2
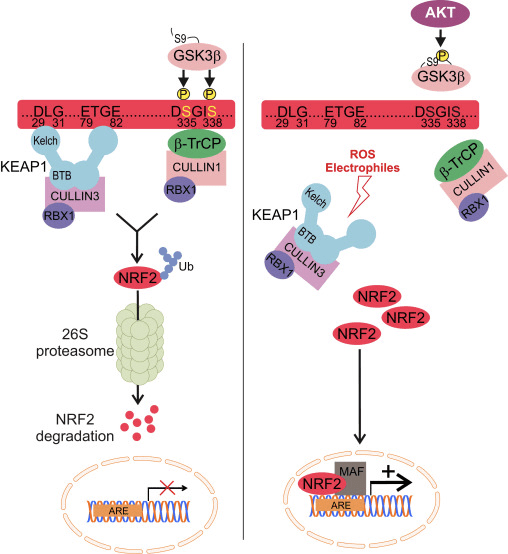
Nrf2 належить до підродини Cap �n� Collar (CNC) і включає сім функціональних доменів, Neh (гомологія Nrf2-ECH) від 1 до Neh7 [9], [10]. Neh1 — це домен CNC-bZIP, який дозволяє Nrf2 гетеродимеризуватися з білком малої м’язовоапоневротичної фібросаркоми (Maf), ДНК та іншими транскрипційними партнерами, а також утворювати ядерний комплекс з ферментом UbcM2, що кон’югує убіквітин [11], [12]. Neh2 містить два важливі мотиви, відомі як DLG і ETGE, які є суттєвими для взаємодії між Nrf2 та його негативним регулятором Keap1 [13], [14].
Keap1 є адаптером субстрату для убіквітин-лігази E3 на основі куліну, який пригнічує транскрипційну активність Nrf2 шляхом убіквітинування та протеасомної деградації в нормальних умовах [15], [16], [17]. Домени KELCH гомодимеру Keap1 зв’язуються з мотивами DLG і ETGE домену Nrf2-Neh2 в цитозолі, де ETGE діє як шарнір з більш високою спорідненістю, а DLG діє як засувка [18]. Під окислювальним стресом або під впливом активаторів Nrf2 Nrf2 відокремлюється від зв’язування Keap1 через тіолову модифікацію залишків цистеїну Keap1, що в кінцевому підсумку запобігає убіквітинації Nrf2 та протеасомній деградації [19]. Потім Nrf2 транслокується в ядро, гетеродимеризується за допомогою невеликих білків Maf і трансактивує батарею генів ARE (рис. 1A). Карбокси-кінець Neh3 діє як домен трансактивації, взаємодіючи з коактиватором транскрипції, відомим як CHD6 (білок, що зв’язує ДНК хромо-АТФази/гелікази) [20]. Neh4 і Neh5 також діють як домени трансактивації, але зв’язуються з іншим коактиватором транскрипції, відомим як CBP (cAMP-response-element-binding protein-binding protein) [21]. Крім того, Neh4 і Neh5 взаємодіють з ядерним кофактором RAC3/AIB1/SRC-3, що призводить до посилення експресії гена ARE, націленого на Nrf2 [22]. Neh5 має окислювально-відновний сигнал ядерного експорту, який має вирішальне значення для регуляції та клітинної локалізації Nrf2 [23].
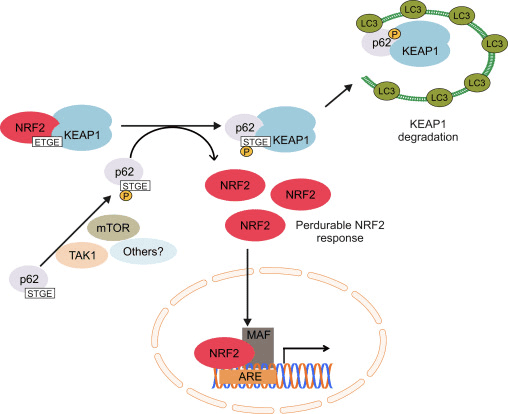
Рисунок 1 Keap1-залежне та -незалежне регулювання Nrf2. (A) У базальних умовах Nrf2 секвеструється з Keap1 двома його мотивами (ETGE і DLG), що призводить до опосередкованого CUL3 убіквітинування з подальшою деградацією протеасоми. Під впливом окисного стресу Nrf2 дисоціює від Keap1, переміщається в ядро і активує батарею ARE-гена. (B) GSK3 фосфорилює Nrf2, і це полегшує розпізнавання Nrf2 ?-TrCP для CUL1-опосередкованого убіквітинування та подальшої деградації протеасоми. (C) p62 секвеструється з Keap1, що призводить до його аутофагічної деградації, вивільнення Nrf2 та посилення передачі сигналів Nrf2.
Keap1-Незалежне регулювання Nrf2
Нові докази виявили новий механізм регуляції Nrf2, який не залежить від Keap1. Багатий серином домен Neh6 Nrf2 відіграє вирішальну роль у цій регуляції, зв’язуючись з двома своїми мотивами (DSGIS і DSAPGS) з білком, що містить повтори ?-трансдуцину (?-TrCP) [24]. ?-TrCP є субстратним рецептором для комплексу убіквітин-лігази Skp1�Cul1�Rbx1/Roc1, який націлений на Nrf2 для убіквітинування та деградації протеасом. Глікогенсинтаза кіназа-3 є важливим білком, який бере участь у Keap1-незалежній стабілізації та регуляції Nrf2; він фосфорилює Nrf2 в домені Neh6, щоб полегшити розпізнавання Nrf2 ?-TrCP і подальшу деградацію білка [25] (рис. 1B).
Інші регулятори Nrf2
Інший ряд доказів виявив неканонічний шлях p62-залежної активації Nrf2, при якому p62 секвеструє Keap1 до аутофагічної деградації, що в кінцевому підсумку призводить до стабілізації Nrf2 і трансактивації Nrf2-залежних генів [26], [27], [ 28], [29] (рис. 1В).
Накопичені дані свідчать про те, що декілька мікроРНК відіграють важливу роль у регуляції активності Nrf2 [30]. Сангокоя та ін. [31] продемонстрували, що miR-144 безпосередньо пригнічує активність Nrf2 у клітинній лінії лімфобласта K562, первинних клітинах-попередниках еритроїду людини та ретикулоцитах серповидно-клітинної хвороби. Інше цікаве дослідження епітеліальних клітин молочної залози людини показало, що miR-28 інгібує Nrf2 через Keap1-незалежний механізм [32]. Аналогічно, мікроРНК, такі як miR-153, miR-27a, miR-142-5p і miR144, знижують експресію Nrf2 в лінії клітин нейрона SH-SY5Y [33]. Сінгх та ін. [34] продемонстрували, що ектопічна експресія miR-93 зменшує експресію Nrf2-регульованих генів у моделі канцерогенезу молочної залози, індукованої 17?-естрадіолом (E2).
Нещодавнє відкриття нашої лабораторії виявило ендогенний інгібітор Nrf2, відомий як ретиноєвий X-рецептор альфа (RXR?). RXR? є ядерним рецептором, взаємодіє з доменом Neh7 Nrf2 (амінокислотні залишки 209×316) через свій ДНК-зв’язуючий домен (DBD) і специфічно інгібує активність Nrf2 в ядрі. Крім того, повідомлялося, що інші ядерні рецептори, такі як рецептор-α, що активується проліфератором пероксисом, ER?, рецептор, пов’язаний з естрогеном-α, і рецептори глюкокортикоїдів, також є ендогенними інгібіторами активності Nrf2 [9], [10].
Протизапальна роль Nrf2/HO-1 Axis
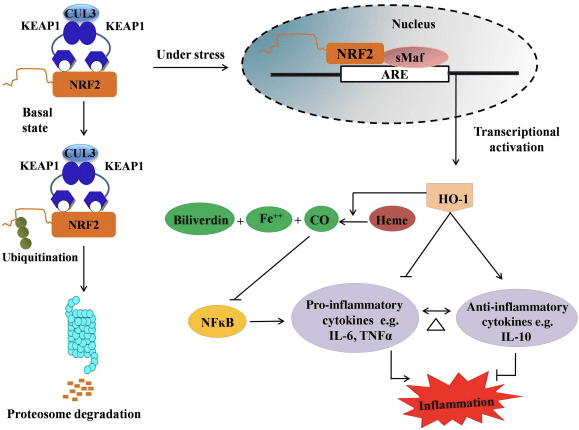
HO-1 — це індукційна ізоформа та фермент, що обмежує швидкість, що каталізує розпад гему до оксиду вуглецю (CO) і вільного заліза, а білівердину — до білірубіну. Ферментативна деградація вільного прозапального гему, а також вироблення протизапальних сполук, таких як CO та білірубін, відіграють важливу роль у підтримці захисних ефектів HO-1 (рис. 2).
Малюнок 2 Огляд шляху Nrf2/HO-1. У базальних умовах Nrf2 зв’язується зі своїм репресором Keap1, що призводить до убіквітинування з наступною деградацією протеасоми. Під час окисного стресу вільний Nrf2 переміщується в ядро, де димерізується з членами невеликого сімейства Maf і зв’язується з генами ARE, такими як HO-1. Підвищена регуляція HO-1 каталізує гем у СО, білірубін і вільне залізо. CO діє як інгібітор шляху NF-?B, що призводить до зниження експресії прозапальних цитокінів, тоді як білірубін також діє як антиоксидант. Крім того, HO-1 безпосередньо інгібує прозапальні цитокіни, а також активує протизапальні цитокіни, таким чином призводить до балансування запального процесу.
Nrf2 індукує ген HO-1 шляхом збільшення експресії мРНК та білка, і це один із класичних регульованих генів Nrf2, який широко використовується у численних дослідженнях in vitro та in vivo. Кілька досліджень продемонстрували, що HO-1 та його метаболіти мають значний протизапальний ефект, опосередкований Nrf2. Підвищення експресії HO-1, що опосередковується активованим Nrf2, призводить до інгібування передачі сигналів NF?B, що призводить до зменшення пошкодження слизової оболонки кишечника та дисфункції щільного з’єднання в моделі трансплантації печінки щура Sprague-Dawley [35]. Підвищення регуляції Nrf2-залежної експресії HO-1 може захистити міобласти C2C12 миші від цитотоксичності H2O2 [36]. Nrf2-залежний HO-1 впливає на опосередковані ліпополісахаридами (LPS) запальні реакції в RAW264.7- або макрофагах пінистих клітин перитонеального макрофага миші. Активність Nrf2 десенсибілізує фенотип пінних клітин макрофагів і запобігає непомірному запаленню макрофагів, які відіграють важливу роль у прогресуванні атеросклерозу [37]. Вісь Nrf2/HO-1 впливає на індуковані LPS мікрогліальні клітини BV2 миші та клітини HT22 гіпокампу миші, впливаючи на нейрозапалення. Підвищення експресії HO-1 через шлях Nrf2 у клітинах мікроглії BV2 миші, які захищають клітинну смерть клітин HT22 гіпокампу миші [38]. Крім того, гібридні молекули на основі кобальту (HYCOs), які поєднують індуктор Nrf2 з вивільнеником монооксиду вуглецю (CO), збільшують експресію Nrf2/HO-1, вивільняють CO та виявляють протизапальну дію in vitro. HYCO також активізують тканинну HO-1 і доставляють CO в кров після введення in vivo, підтримуючи їх потенційне використання проти запальних станів [39]. Підвищення регуляції Nrf2/HO-1 зменшує запалення за рахунок збільшення ефероцитарної активності мишачих макрофагів, які обробляються тауриновими хлорамінами [40]. Загалом, роз’яснені вище експериментальні моделі показали, що вісь Nrf2/HO-1 відіграє важливу роль у протизапальній функції, що свідчить про те, що Nrf2 є терапевтичною мішенню при захворюваннях, пов’язаних із запаленням.
Крім того, побічні продукти HO-1, такі як CO, білірубін, діє як потужний антиоксидант під час окисного стресу та пошкодження клітин [41], [42]; пригнічує аутоімунний енцефаломієліт та гепатит [43], [44]; і він захищає мишей і щурів від ендотоксичного шоку, запобігаючи утворення iNOS і NO [45], [46], [47]. Більше того, білірубін зменшує активацію ендотелію та дисфункцію [48]. Цікаво, що білірубін зменшує трансміграцію ендотеліальних лейкоцитів через молекулу адгезії-1 [49]. Ці конкретні посилання вказують, що не тільки HO-1 діє як потужний протизапальний засіб, але й його метаболіти.
Медіатори запалення та ферменти, інгібовані Nrf2
Цитокіни та хемокіни
Цитокіни — це низькомолекулярні білки та поліпептиди, що виділяються різними клітинами; вони регулюють ріст клітин, диференціацію та імунну функцію, а також беруть участь у запаленні та загоєнні ран. Цитокіни включають інтерлейкіни (IL), інтерферони, фактор некрозу пухлин (TNF), колонієстимулюючий фактор, хемокіни та фактори росту. Деякі цитокіни вважаються прозапальними медіаторами, тоді як інші мають протизапальну функцію. Вплив окисного стресу призводить до надлишкового виробництва цитокінів, що викликає окислювальний стрес у клітинах-мішенях. Кілька прозапальних цитокінів надлишково виробляються, коли NF-?B активується окислювальним стресом. Крім того, прозапальний окислювальний стрес викликає подальшу активацію NF-?B і надлишкове виробництво цитокінів. Активація системи Nrf2/ARE відіграє важливу роль у порушенні цього циклу. Хемокіни — це сімейство невеликих цитокінів, головна роль яких полягає в управлінні міграцією запальних клітин. Вони діють переважно як хемоатрактанти для лейкоцитів, моноцитів, нейтрофілів та інших ефекторних клітин.
Повідомлялося, що активація Nrf2 запобігає індукованій LPS транскрипційної регуляції прозапальних цитокінів, включаючи IL-6 та IL-1? [50]. ІЛ-1? і виробництво IL-6 також збільшується в Nrf2?/? миші з декстрансульфат-індукованим колітом [51], [52]. Nrf2 пригнічує продукцію IL-17 та інших факторів запалення Th1 і Th17, а також пригнічує процес захворювання в експериментальній моделі розсіяного склерозу, аутоімунного енцефаліту [53]. Nrf2-залежні антиоксидантні гени HO-1, NQO-1, Gclc і Gclm блокують TNF-?, IL-6, моноцитний хіміоатрактантний білок-1 (MCP1), макрофагальний запальний білок-2 (MIP2) і запальний посередники. Але у випадку Nrf2-нокаутних мишей протизапальний ефект не виникає [54]. Перитонеальні нейтрофіли від мишей з нокаутом Nrf2, які отримували ЛПС, мають значно вищі рівні цитокінів (TNF-? та IL-6) та хемокінів (MCP1 та MIP2), ніж клітини дикого типу (WT) [54]. In vitro, перенесення гена Nrf2 до гладких м’язових клітин аорти людини та кролика пригнічує секрецію MCP1 [8], [55], а експресія Nrf2-залежної HO-1 пригнічує TNF-?-стимульовані NF-?B і MCP-1 секреція в ендотеліальних клітинах пупкової вени людини [56]. Ці результати натякають на те, що у відповідь на запальні стимули посилення передачі сигналів Nrf2 пригнічує надлишкову продукцію прозапальних цитокінів і хемокінів, а також обмежує активацію NF-?B.
Молекули клітинної адгезії
Молекули клітинної адгезії (CAM) — це білки, які зв’язуються з клітинами або з позаклітинним матриксом. Розташовані на поверхні клітини, вони беруть участь у розпізнаванні клітин, їх активації, передачі сигналу, проліферації та диференціації. Серед CAM, ICAM-1 і VCAM-1 є важливими членами суперсімейства імуноглобулінів. ICAM-1 присутній у низьких концентраціях у мембранах лейкоцитів та ендотеліальних клітин. При стимуляції цитокінами концентрація значно підвищується. ICAM-1 може індукуватися IL-1 і TNF і експресується ендотелієм судин, макрофагами та лімфоцитами. Це ліганд для інтегрину, рецептора, знайденого на лейкоцитах. Коли ICAM-1-інтегриновий міст активується, лейкоцити зв’язуються з ендотеліальними клітинами, а потім мігрують у субендотеліальні тканини [57]. VCAM-1 опосередковує адгезію лімфоцитів, моноцитів, еозинофілів і базофілів до ендотелію судин і сприяє залученню лейкоцитів, що в кінцевому підсумку призводить до пошкодження тканин через окислювальний стрес. Nrf2 інгібує промоторну активність VCAM-1 [58]. Нижній ген HO-2, регульований Nrf1, може впливати на експресію E-селектину та VCAM-1, молекул адгезії, пов’язаних з ендотеліальними клітинами [59]. Легенева експресія кількох CAM, таких як CD-14, TREM1, SELE, SELP і VCAM-1, значно вище в Nrf2?/? мишей, ніж у мишей Nrf2+/+ [60]. Nrf2 в ендотеліальних клітинах аорти людини пригнічує експресію VCAM-1, індуковану TNF-?, і перешкоджає адгезії моноцитарних клітин U937, індукованої TNF-? [8]. Надекспресія Nrf2 також інгібує експресію гена VCAM-1, індукованої TNF-?, у клітинах мікросудинного ендотелію людини [61]. Встановлено, що природний антиоксидант 3-гідроксиантранілова кислота (ГК), один з метаболітів l-триптофану, що утворюються in vivo по метаболічному шляху, відомому як шлях кінуреніну під час запалення або інфекції, індукує експресію HO-1 і стимулює Nrf2 в пупковій порожнині людини. ендотеліальні клітини вен (HUVECs). Nrf2-залежна експресія HO-1, індукована HA, інгібує секрецію MCP-1, експресію VCAM-1 і активацію NF-kB, пов’язану з пошкодженням і запаленням судин при атеросклерозі [56]. Протипроліферативне та протизапальне синтетичне похідне халкону 2?,4?,6?-трис (метоксиметокси) халкону інгібує ICAM-1, прозапальний цитокін IL-1? і TNF-? експресія в тканині товстої кишки у мишей, які отримували тринітробензолсульфонову кислоту [62]. Підвищення регуляції Nrf2 інгібує TNF-?-індуковану експресію ICAM-1 у пігментних епітеліальних клітинах сітківки сітківки людини, оброблених лікопіном [63]. Усі ці дослідження показують, що Nrf2 відіграє ключову роль у запальному процесі, регулюючи міграцію та інфільтрацію запальних клітин до запаленої тканини.
Матричні металопротеїнази (ММР)
ММР широко присутні в позаклітинному матриксі і беруть участь у фізіологічних і патологічних процесах, таких як клітинна проліферація, міграція, диференціація, загоєння ран, ангіогенез, апоптоз і метастазування пухлин. Повідомлялося, що вісь Nrf2/HO-1 інгібує ММР-9 у макрофагах і ММР-7 в епітеліальних клітинах кишечника людини, і це корисно при лікуванні запальних захворювань кишечника [62], [64]. Ушкодження шкіри, викликане УФ-опроміненням, є більш серйозним у Nrf2-нокауту, ніж у мишей WT, а рівень MMP-9 значно вищий, що вказує на те, що Nrf2 знижує експресію MMP-9. Тому Nrf2 вважається захисним проти УФ-опромінення [65]. В іншому дослідженні також повідомляється, що активація транскрипційної активації MMP-9 при інвазії та запаленні пухлинних клітин регулюється шляхом інгібування сигнального шляху NF-kB [66]. При травматичному ураженні спинного мозку сигнальний шлях NF-kB також бере участь у регулюванні рівнів мРНК ММР-9 [67]. Таким чином, при запаленні на регуляцію MMP впливає безпосередньо шлях Nrf2 або опосередковано через NF-?B шлях під впливом Nrf2.
Циклооксигеназа-2 (ЦОГ2) та індукційна синтаза оксиду азоту (INOS)
Серія експериментів на мишах з нокаутом Nrf2 продемонструвала його вирішальну роль у запаленні та регуляції прозапальних генів, таких як COX-2 та iNOS. Вперше Хор та ін. повідомляють про підвищену експресію прозапальних цитокінів, таких як ЦОГ-2 та iNOS, у тканинах товстої кишки Nrf2?/? мишей порівняно з мишами WT Nrf2+/+, що вказує на те, що Nrf2 пригнічує їхню активність [51]. Інша доповідь про попередню обробку сульфорафаном, одним із добре відомих активаторів Nrf2, присутніх в овочах хрестоцвітих, продемонструвала його протизапальну дію, інгібуючи експресію TNF-?, IL-1?, ЦОГ-2 та iNOS на обох мРНК. і рівні білка в первинних перитонеальних макрофагах мишей Nrf2+/+ порівняно з макрофагами Nrf2?/? мишей [68]. Аналогічно, гіпокамп мишей з нокаутом Nrf2 із запаленням, спричиненим ЛПС, також демонструє більш високу експресію маркерів запалення, таких як iNOS, IL-6 та TNF-? ніж мишей WT [69]. Подібним чином миші з Nrf2-нокаутом гіперчутливі до окисного стресу, викликаного 1-метил-4-феніл-1,2,3,6-тетрагідропіридином, а також демонструють підвищені рівні мРНК і білка маркерів запалення, таких як ЦОГ-2, iNOS. , ІЛ-6 та TNF-? [70]. Більше того, печінки з Nrf2?/? у мишей, які страждали на дієту з дефіцитом метіоніну та холіну, експресія мРНК Cox5 та iNOS приблизно в 2 разів вища, ніж у мишей WT на тій же дієті, що свідчить про протизапальну роль Nrf2 [71]. Нещодавно Kim et al. продемонстрували, що фітохімічний етилпіруват виявляє протизапальну та антиоксидантну дію, зменшуючи експресію iNOS через передачу сигналів Nrf2 в клітинах BV2. Вони показали, що етилпіруват індукує ядерну транслокацію Nrf2, яка в кінцевому підсумку пригнічує взаємодію між p65 і p300, що призводить до зниження експресії iNOS [72]. Крім того, аналог карбазолу LCY-2-CHO активує Nrf2 і викликає його ядерну транслокацію, що призводить до пригнічення експресії COX2 та iNOS [73] в гладком’язових клітинах судин аорти щурів.
Парадоксальна роль Nrf2 в регуляції активності Iflammasome NLRP3
Сімейство NLR, піриновий домен, що містить 3 (NLRP3) інфламасома, є мультибілковим комплексом, який функціонує як рецептор розпізнавання патогенів (PRR) і розпізнає широкий спектр мікробних сигналів окисного стресу, таких як патоген-асоційовані молекулярні структури (PAMP), пошкодження- пов'язані молекули молекулярного малюнка (DAMP) і АФК [74]. Активована інфламасома NLRP3 опосередковує розщеплення каспази-1 і секрецію прозапального цитокіну інтерлейкіну-1? (IL-1?), що в кінцевому підсумку індукує процес загибелі клітин, відомий як піроптоз, який захищає господаря від широкого кола патогенів [75]. Однак аберантна активація інфламасоми пов’язана із захворюваннями неправильного згортання білків, такими як трансмісивна губчаста енцефалопатія, хвороба Альцгеймера, хвороба Паркінсона, а також цукровий діабет 2 типу [76], рак [77], подагра та атеросклероз [78].
Нещодавнє спостереження групи Rong Hu щодо асоціації Nrf2 з негативною регуляцією запалення показало, що Nrf2 індукує експресію NQO1, що призводить до інгібування активації інфламасоми NLRP3, розщеплення каспази-1 та IL-1? утворення в макрофагах. Крім того, добре відомий активатор Nrf2, трет-бутилгідрохінон (tBHQ), негативно регулював транскрипцію NLRP3, активуючи ARE Nrf2-залежним способом [79]. На додаток до вищенаведеного спостереження, у цій же групі також було виявлено, що диметилфумарат (DMF) запобігає DSS-індукований коліт шляхом активації сигнального шляху Nrf2, який бере участь у транслокації ядер Nrf2 та інгібуванні зборки інфламасоми NLRP3 [80].
Серія експериментів із використанням природних і синтетичних сполук також виявила інгібуючий ефект Nrf2 на активацію інфламасоми NLRP3. Наприклад, лікування епігалокатехін-3-галлатом (EGCG) у мишей з вовчаковим нефритом показало зниження активації ниркової інфламасоми NLRP3, що опосередковується сигнальним шляхом Nrf2 [81]. Аналогічно, цитраль (3,7-диметил-2,6-октадієнал), головна активна сполука в китайському фітопрепараті Litsea cubeba, інгібує активацію інфламасоми NLRP3 через антиоксидантний сигнальний шлях Nrf2 у моделі прискореного та важкого вовчакового нефриту (ASLN). [82]. Аналогічно, біоханін захищав від ураження печінки, спричиненого LPS/GalN, активуючи шлях Nrf2 та інгібуючи активацію інфламасоми NLRP3 у мишей-самців BALB/c [83]. Крім того, було показано, що мангіферин посилює експресію Nrf2 і HO-1 залежно від дози та інгібує LPS/D-GalN-індукований печінковий NLRP3, ASC, каспазу-1, IL-1? і TNF-? вираз [84].
Незважаючи на негативну регуляцію NLRP3 Nrf2, він також активує функцію запалення NLRP3 і AIM2. Хайтао Вен і його колеги виявили, що Nrf2 ?/? макрофаги мишей продемонстрували дефектну активацію інфламасоми NLRP3 і AIM2, але не інфламасоми NLRC4 [85]. Цікаво, що це спостереження зображує невідомі функції Nrf2 у контексті захворювань, пов’язаних із запаленням; тому дуже важливо дослідити далі, щоб виявити механізм, за яким Nrf2 активує функцію інфламасоми, перш ніж розглядати її як терапевтичну мішень.
Нещодавнє дослідження, засноване на результатах імунопреципітації хроматину (ChIP)-seq і ChIP-qPCR у макрофагах мишей, показало, що Nrf2 зв’язується з промоторними ділянками прозапальних цитокінів, таких як IL-6 та IL-1? і інгібує залучення РНК Pol II. В результаті РНК Pol II не в змозі обробити транскрипційну активацію IL-6 та IL-1? що в кінцевому підсумку призводить до пригнічення експресії генів. Вперше група Масаюкі Ямамото розкрила новий механізм, за допомогою якого Nrf2 не тільки трансактивує свої гени, що знаходяться нижче по ходу за допомогою ARE, але й пригнічує транскрипційну активацію специфічних генів з або без ARE шляхом інгібування залучення РНК Pol II [50].
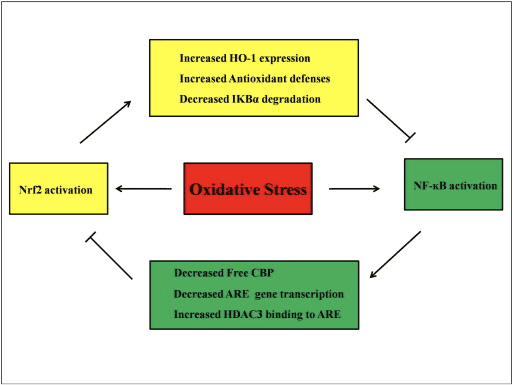
Перехресні перешкоди між шляхами Nrf2 і NF-?B
NF-?B — це білковий комплекс, відповідальний за транскрипцію ДНК, який зустрічається майже в усіх типах клітин тварин і бере участь у різних процесах, таких як запалення, апоптоз, імунна відповідь, ріст і розвиток клітин. p65, білок Rel сімейства NF-?B, має домен трансактивації, тоді як p50 його не має і вимагає гетеродимеризації білком Rel для активації транскрипції. Під час окисного стресу активується I?B кіназа (IKK) і викликає фосфорилювання I?B, що призводить до вивільнення та ядерної транслокації NF-?B. NF-?B викликає транскрипцію прозапальних медіаторів, таких як IL-6, TNF-?, iNOS, IL-1 і внутрішньоклітинної адгезії ЦОГ-2.
Аномальна регуляція NF-?B була пов’язана з ревматоїдним артритом, астмою, запальним захворюванням кишечника та гастритом, спричиненим інфекцією Helicobacter pylori [86]. В даний час вважається, що активність NF-kB впливає на сигнальний шлях Keapl/Nrf2/ARE головним чином у трьох аспектах: по-перше, Keap1 руйнує IKK? шляхом убіквітинування, таким чином пригнічуючи активність NF-?B [87]. По-друге, запальний процес індукує медіатори запалення, такі як COX2, отримані з циклопентенону простагландину 15d-PGJ2, сильного електрофіла, який реагує з Keap1 і активує Nrf2, таким чином ініціюючи транскрипцію гена з одночасним пригніченням активності NF-kB [58] [88] Рис. 3 A, B). По-третє, NF-?B може поєднуватися з конкурентним коактиватором транскрипції Nrf2 CBP [89], [90] (рис. 3 C, D).
Малюнок 3 Перехресні перешкоди між шляхами Nrf2 і NF-?B. (A) Keap1 спрямовує IKK на CUL3-опосередковане убіквітинування та деградацію протеасом, що в кінцевому підсумку призводить до інгібування фосфорилювання NF-?B, і цей механізм також працює як конкурентне зв'язування Nrf2 і IKK з Keap1. (B) Окислювальний стрес активує IKK, який фосфорилює NF-?B, що призводить до його транслокації в ядро та активації прозапальних цитокінів, таких як ЦОГ-2. Кінцевий продукт ЦОГ-2, відомий як 15d-PGJ2, діє як індуктор Nrf2, що в кінцевому підсумку призводить до придушення окисного стресу. (C) Nrf2 зв'язується зі своїм транскрипційним кофактором CBP разом з невеликим Maf та іншими транскрипційними механізмами, щоб ініціювати експресію гена, керовану ARE. (D) Коли NF-?B зв'язується з CBP у конкурентний спосіб, він інгібує зв'язування CBP з Nrf2, що призводить до інгібування трансактивації Nrf2.
Передбачається, що сигнальні шляхи Nrf2 і NF-?B взаємодіють, щоб контролювати транскрипцію або функцію нижніх цільових білків. На підтвердження цього припущення багато прикладів показують, що пряма або непряма активація та інгібування відбуваються між членами шляхів Nrf2 і NF-?B (рис. 4). У відповідь на LPS, нокдаун Nrf2 значно збільшує транскрипційну активність NF-?B і NF-?B-залежну транскрипцію гена, показуючи, що Nrf2 перешкоджає активності NF-?B [60], [91]. Крім того, підвищена експресія Nrf2-залежної HO-1 пригнічує активність NF-?B. Коли клітини раку передміхурової залози короткочасно піддаються впливу ?-тохоферилсукцинату, похідного вітаміну Е, експресія HO-1 посилюється. Кінцеві продукти HO-1 інгібують ядерну транслокацію NF-?B [92]. Ці дослідження in vivo свідчать про те, що Nrf2 негативно регулює сигнальний шлях NF-kB. LPS стимулює активність зв'язування ДНК NF-?B, і рівень субодиниці p65 NF-?B значно вище в ядерних екстрактах з легенів Nrf2?/? ніж від мишей WT, що свідчить про негативну роль Nrf2 в активації NF-?B. Крім того, Nrf2?/? Фібробласти ембріонів миші, оброблені LPS та TNF-? показують більш помітну активацію NF-?B, викликану активацією IKK і I?B-? деградація [60]. І кліренс респіраторно-синцитіального вірусу значно зменшується, тоді як NF-?B ДНК-зв'язуюча активність збільшується в Nrf2?/? мишей порівняно з мишами WT [93]. Пристан-індукований вовчаковий нефрит у Nrf2?/? миші, які одночасно отримували сульфорафан, мають серйозне ураження нирок і патологічні зміни, а також підвищену експресію iNOS і активацію NF-?B порівняно з WT, що свідчить про те, що Nrf2 покращує вовчаковий нефрит шляхом інгібування сигнального шляху NF-?B і очищення АФК [94 ]. Активність NF-?B також виникає, коли клітини обробляють індуктором Nrf2 разом з LPS і TNF-?. Наприклад, синтетичне похідне халкону інгібує TNF-?-індуковану активацію NF-?B як прямо, так і опосередковано, а також частково через індукцію експресії HO-1 в епітеліальних клітинах кишечника людини HT-29 [62]. Пригнічення транслокації NF-?B і ДНК-зв’язування активності, а також пригнічення експресії iNOS в гепатоцитах виявлено, коли щурів F344 обробляли 3H-1,2-дитіол-3-тіоном (D3T) [95]. Після спільного лікування сульфорафаном і ЛПС, ЛПС-індукована експресія iNOS, ЦОГ-2 і ФНП-? у Raw 264.7 макрофагів знижується регуляція, що дозволяє припустити, що сульфорафан має протизапальну активність через пригнічення зв’язування NF-?B ДНК [96]. Хоча було проведено кілька експериментальних досліджень, щоб пояснити зв’язок між шляхами Nrf2 і NF-?B, результати залишаються суперечливими. Повідомлялося про позитивну і негативну регуляцію між Nrf2 і NF-kB [97]. Зазвичай хіміопрофілактичні електрофіли 3H-1,2-дитіол-3-тіон, сульфорафан і тритерпеноїд CDDO-Me активують Nrf2 шляхом інгібування NF-kB та його генів, що знижуються [98], [99], [100]. На противагу цьому, було показано, що кілька агентів або умов, таких як ROS, LPS, напруга зсуву потоку, окислений ЛПНЩ і сигаретний дим, підвищують активність як Nrf2, так і NF-kB [97]. Крім того, дослідження in vivo показали, що активність NF-kB знижена в печінці, виділеній з Nrf2?/? мишей і активність зв'язування NF-?B нижча в Nrf2?/? ніж у мишей Nrf2+/+ [101]. Проте ендотеліальні клітини аорти людини, оброблені аденовірусним вектором Nrf2, інгібують нижні гени NF-?B, не впливаючи на активність NF-?B [8].
Рисунок 4 Регуляторний контур Nrf2 і NF-?B. Шлях Nrf2 інгібує активацію NF-?B, запобігаючи деградації I?B-? і підвищення експресії HO-1 і антиоксидантного захисту, які нейтралізують АФК і детоксикаційні хімічні речовини. В результаті активація NF-?B, пов'язана з ROS, пригнічується. Аналогічно, NF-?B-опосередкована транскрипція зменшує активацію Nrf2, зменшуючи�ЕСТЬТранскрипція гена та вільний білок, що зв’язує CREB, конкуруючи з Nrf2 за CBP. Більше того, NF-?B збільшує залучення гістондеацетилази (HDAC3) в область ARE, і, отже, активація транскрипції Nrf2 запобігається.
Активація сигнального шляху Nrf2 відіграє важливу роль у експресії ферментів і генів, які беруть участь у детоксикації реактивних окислювачів шляхом підвищення антиоксидантної здатності клітин в організмі людини. Хоча сьогодні доступно багато досліджень, регуляторні механізми активації Nrf2 не повністю зрозумілі. Також виявлено можливу роль сигнального шляху Nrf2 у лікуванні запалення. Д-р Алекс Хіменес, округ Колумбія, CCST Insight
Роль Nrf2 у запальних захворюваннях
Дослідження in vivo показали, що Nrf2 відіграє важливу роль у запальних захворюваннях, що вражають різні системи; до них належать гастрит, коліт, артрит, пневмонія, ураження печінки, серцево-судинні захворювання, нейродегенеративні захворювання та ураження мозку. У цих дослідженнях Nrf2?/? тварини виявляли більш серйозні симптоми запалення та пошкодження тканин, ніж тварини WT. Тому вважається, що сигнальний шлях Nrf2 має захисну дію при запальних захворюваннях. Інтратрахеальна установка панкреатичної еластази свиней викликає хронічну обструктивну хворобу легень, зокрема емфізему. Миші з дефіцитом Nrf2 дуже сприйнятливі до емфіземи, а в альвеолярних макрофагах спостерігається зниження експресії HO-1, PrxI та антипротеазного гена SLPI. Nrf2 вважається ключовим регулятором у системі захисту, опосередкованої макрофагами, проти ураження легень [102]. Миші з дефіцитом Nrf2 з емфіземою, спричиненою впливом тютюнового диму протягом 6 місяців, демонструють посилене бронхоальвеолярне запалення, підвищену експресію маркерів окисного стресу в альвеолах та збільшений апоптоз клітин альвеолярної перегородки, що свідчить про те, що Nrf2 діє проти тютюнового диму через посилення експресії емоксиду, індукованого тютюном. гени [102], [103]. При порушенні Nrf2 алерген-опосередковане запалення дихальних шляхів і астма за допомогою комплексу овалбуміну показують підвищене запалення дихальних шляхів, гіперреактивність дихальних шляхів, гіперплазію келихоподібних клітин і високий рівень Th2 в бронхоальвеолярному змиві та спленоцитах, тоді як Nrf2-опосередкований сигнальний шлях дихальних шляхів обмежує сигнальні шляхи дихальних шляхів. , гіперсекреція слизу та гіперреактивність дихальних шляхів, а також індукування багатьох антиоксидантних генів, які запобігають розвитку астми [104]. Ін’єкція каррагінану в плевральну порожнину викликає плеврит, і накопичення 15d-PGJ2 в запальних клітинах Nrf2 обмежується перитонеальними макрофагами миші. Під час ранньої фази запалення 15d-PGJ2 активує Nrf2 і регулює запальний процес шляхом індукції HO-1 і PrxI. Дослідження також показало, що ЦОГ-2 має протизапальну дію на ранній фазі шляхом вироблення 15d-PGJ2 [105]. Пероральний прийом 1% декстрансульфату натрію протягом 1 тижня викликає коліт, пов’язаний з гістологічними змінами, які включають вкорочення крипт та інфільтрацію запальних клітин у тканині товстої кишки. Для захисту цілісності кишечника при коліті Nrf2 може відігравати важливу роль, регулюючи прозапальні цитокіни та індукуючи ферменти фази II детоксикації [51]. У моделі миші з нокаутом Nrf2 LPS-індукованого легеневого сепсису активність NF-?B регулює вплив запальних цитокінів, таких як ЦОГ-2, IL-113, IL-6 і TNF? які є важливими для ініціації та сприяння запалення [60]. Nrf2 зменшує запальні пошкодження, регулюючи ці фактори запалення. У цих моделях гострого запалення підвищена регуляція антиоксидантних ферментів, прозапальних цитокінів і медіаторів за допомогою сигнального шляху Nrf2 зменшує запальне ураження у тварин WT. Цікаво, що про це також повідомлялося у мишей з нокаутом Nrf2, у яких симптоми помітно посилюються порівняно з мишами WT.
Дослідження Nrf2-залежних протизапальних препаратів
Підсумовуючи, ми обговорили експерименти, які показують, що сигнальний шлях Nrf2 відіграє регулюючу роль у багатьох областях запалення, тому Nrf2-залежні протизапальні засоби є важливими для лікування запальних захворювань.
Рослини були надзвичайно багатими джерелами сполук, які активують фактор транскрипції Nrf2, що призводить до активізації цитопротекторних генів. Нещодавно було проведено кілька досліджень з метою вивчення дії різних протизапальних засобів, переважно рослинного походження. Наприклад, куркумін є активним інгредієнтом куркуми, а також у невеликих кількостях міститься в імбирі; ізотіоціанати, зокрема фенілізотіоціанати, отримують з брокколі, селери та інших овочів; а антоціани – з ягід і винограду [124]. Дослідження показали, що всі ці агенти є не тільки хорошими антиоксидантами, але й мають потужну протизапальну дію через індукцію Nrf2 [125], [126]. Тому розробка нових протизапальних активаторів Nrf2 з рослинного екстракту викликала великий інтерес у медичних дослідженнях.
В останні роки було проведено багато експериментів на тваринах для підтвердження дії цих сполук. Артесунат використовується в основному при важкій малярії, церебральній малярії та ревматичних аутоімунних захворюваннях; він також ефективний при септичному ураженні легенів. Артесунат активує експресію Nrf2 і HO-1, а остання зменшує надходження прозапальних цитокінів і лейкоцитів у тканини для запобігання запалення [127]. Вважається, що ізовітексин, витягнутий з лушпиння рису Oryza sativa, має протизапальні та антиоксидантні властивості; він відіграє захисну роль від гострого ураження легень, спричиненого ЛПС, активуючи шлях Nrf2/HO-1 та інгібуючи MAPK та NF-?B [128]. Фімасартан, нещодавно популярний блокатор рецепторів ангіотензину II, що діє на систему ренін-ангіотензин, знижує артеріальний тиск; Використання фімасартану для лікування мишей з односторонньою обструкцією сечоводу, викликаною хірургічним шляхом, зменшує окислювальний стрес, запалення та фіброз за рахунок активізації Nrf2 та антиоксидантного шляху та інгібування RAS та MAPK [129]. Саппанон широко поширений у Південно-Східній Азії, де його використовують як протигрипозний, протиалергічний і нейропротекторний препарат; він активує Nrf2 і інгібує NF-?B, і тому може бути корисним при лікуванні захворювань, пов'язаних з Nrf2- та/або NF-?B [130]. Біксин, отриманий з насіння Bixin orellana, використовується для лікування інфекційно-запальних захворювань у Мексиці та Південній Америці; він зменшує медіатори запалення, витікання альвеолярних капілярів та окислювальне пошкодження залежно від Nrf2, щоб полегшити спричинене вентиляцією пошкодження легень та відновити нормальну морфологію легень [131]. Інші рослинні сполуки, такі як епігалокатехін галлат, сульфорафан, ресвератрол, лікопін та екстракт зеленого чаю, мають терапевтичний вплив на запальні захворювання через сигнальний шлях Nrf2 [132], [133], [134]. Нещодавно було повідомлено, що інший фітохімічний препарат, ериодиктиол, який присутній у цитрусових, має протизапальну та антиоксидантну дію на спричинене цисплатином ушкодження нирок та спричинене сепсисом гостре ушкодження легенів шляхом регулювання Nrf2, інгібування NF-?B та інгібування експресія цитокінів у макрофагах [135], [136]. Однак численні фітохімічні речовини мають великі перспективи для профілактики та лікування різних захворювань людини, а деякі вже увійшли в стадію клінічних випробувань (Таблиця 2).
Ці рослинні сполуки активують сигнальний шлях Nrf2 переважно у вигляді електрофільних матеріалів, які модифікують залишки цистеїну Keap1, що призводить до зв’язування вільного ядерного Nrf2 з ARE, що призводить до активації транскрипції відповідного гена.
Сульфорафан та його вплив на рак, смертність, старіння, мозок і поведінку, хвороби серця тощо
Ізотіоціанати є одними з найважливіших рослинних сполук, які ви можете отримати у своєму раціоні. У цьому відео я роблю для них найповнішу справу, яку коли-небудь робили. Короткий період уваги? Перейдіть до улюбленої теми, натиснувши один із моментів часу нижче. Повна хронологія нижче.
Ключові розділи:
00:01:14 – Рак і смертність
00:19:04 – Старіння
00:26:30 – Мозок і поведінка
00:38:06 – Підсумок
00:40:27 – Доза
Повний графік:
00:00:34 – Представлення сульфорафану, головна тема відео.
00:01:14 – Споживання овочів хрестоцвітних і зниження смертності від усіх причин.
00:02:12 – Ризик раку передміхурової залози.
00:02:23 – Ризик раку сечового міхура.
00:02:34 – Ризик раку легенів у курців.
00:02:48 – Ризик раку молочної залози.
00:03:13 – Гіпотетична: що робити, якщо у вас уже рак? (інтервенційний)
00:03:35 – Імовірний механізм, який керує асоціативними даними раку та смертності.
00:04:38 – Сульфорафан і рак.
00:05:32 – Докази на тваринах, що показують сильний вплив екстракту паростків брокколі на розвиток пухлин сечового міхура у щурів.
00:06:06 – Вплив прямого прийому сульфорафану у пацієнтів з раком передміхурової залози.
00:07:09 – Біоакумуляція метаболітів ізотіоціаната у фактичній тканині молочної залози.
00:08:32 – Пригнічення стовбурових клітин раку молочної залози.
00:08:53 – Урок історії: ще в Стародавньому Римі стверджували, що капустяні гриби мають оздоровчі властивості.
00:09:16 – Здатність сульфорафану посилювати виведення канцерогену (бензолу, акролеїну).
00:09:51 – NRF2 як генетичний перемикач через елементи антиоксидантної реакції.
00:10:10 – Як активація NRF2 посилює виведення канцерогену через глутатіон-S-кон'югати.
00:10:34 – Брюссельська капуста підвищує глутатіон-S-трансферазу і зменшує пошкодження ДНК.
00:11:20 – Напій з проростків брокколі збільшує виведення бензолу на 61%.
00:15:45 – Споживання хрестоцвітних овочів і смертність від серцево-судинних захворювань.
00:16:55 – порошок паростків брокколі покращує рівень ліпідів у крові та загальний ризик серцевих захворювань у діабетиків 2 типу.
00:19:04 – Початок секції старіння.
00:19:21 – Дієта, збагачена сульфорафаном, збільшує тривалість життя жуків від 15 до 30% (за певних умов).
00:20:34 – Важливість слабкого запалення для довголіття.
00:22:05 – Овочі хрестоцвітних і порошок паростків брокколі, здається, зменшують широкий спектр запальних маркерів у людей.
00:23:40 – Підсумок у середині відео: розділи про рак, старіння
00:24:14 – Дослідження на мишах показують, що сульфорафан може покращити адаптивну імунну функцію в літньому віці.
00:25:18 – Сульфорафан покращив ріст волосся у мишачої моделі облисіння. Зображення на 00:26:10.
00:26:30 – Початок розділу «Мозок і поведінка».
00:27:18 – Вплив екстракту паростків брокколі на аутизм.
00:27:48 – Вплив глюкорафаніну на шизофренію.
00:28:17 – Початок обговорення депресії (правдоподібний механізм та дослідження).
00:31:21 – Дослідження на мишах з використанням 10 різних моделей депресії, викликаної стресом, показало, що сульфорафан так само ефективний, як і флуоксетин (прозак).
00:32:00 – Дослідження показує, що пряме вживання глюкорафаніну мишами так само ефективне для запобігання депресії через модель стресу соціальної поразки.
00:33:01 – Початок відділу нейродегенерації.
00:33:30 – Сульфорафан і хвороба Альцгеймера.
00:33:44 – Сульфорафан і хвороба Паркінсона.
00:33:51 – Сульфорафан і хвороба Хантінгтона.
00:34:13 – Сульфорафан збільшує кількість білків теплового шоку.
00:34:43 – Початок секції черепно-мозкової травми.
00:35:01 – Сульфорафан, введений відразу після ЧМТ, покращує пам’ять (дослідження на мишах).
00:35:55 – Сульфорафан і нейрональна пластичність.
00:36:32 – Сульфорафан покращує навчання на моделі діабету ІІ типу у мишей.
00:37:19 – Сульфорафанова і м’язова дистрофія Дюшенна.
00:37:44 – Інгібування міостатину в клітинах-супутниках м’язів (in vitro).
00:38:06 – Пізнє відео: смертність і рак, пошкодження ДНК, окислювальний стрес і запалення, виділення бензолу, серцево-судинні захворювання, діабет ІІ типу, вплив на мозок (депресія, аутизм, шизофренія, нейродегенерація), шлях NRF2.
00:40:27 – Думки щодо визначення дози паростків брокколі або сульфорафану.
00:41:01 – Анекдоти про проростання в домашніх умовах.
00:43:14 – Про температуру приготування та активність сульфорафану.
00:43:45 – Перетворення сульфорафану з глюкорафаніну кишковими бактеріями.
00:44:24 – Добавки працюють краще в поєднанні з активною мирозиназою з овочів.
00:44:56 – Техніка приготування та овочі хрестоцвітних.
00:46:06 – Ізотіоціанати як зоб.
Висновки
В даний час багато досліджень зосереджені на ролі сигнального шляху Nrf2/Keap1/ARE у запаленні. Серед ферментів, які підвищуються під впливом Nrf2, HO-1 є одним із репрезентативних ферментів реакції на стрес. HO-1 має виражені протизапальні та антиоксидантні властивості. Загалом, сигнальний шлях Nrf2 також негативно регулює цитокіни, рилізинг-фактори хемокінів, MMP та інші медіатори запалення COX-2 та iNOS, які прямо чи опосередковано впливають на відповідні шляхи NF-kB та MAPK та інші мережі, які контролюють запалення. Припускається, що сигнальні шляхи Nrf2 і NF-?B взаємодіють, щоб регулювати транскрипцію або функцію нижніх цільових білків. Придушення або інактивація NF-?B-опосередкованої транскрипційної активності через Nrf2, швидше за все, відбувається на ранній фазі запалення, оскільки NF-?B регулює синтез de novo низки прозапальних медіаторів. Однак у дослідженні все ще є деякі обмеження, наприклад, чи існують зв’язки між Nrf2 та іншими сигнальними шляхами, такими як JAK/STAT, значення поточних активаторів Nrf2, отриманих з природних рослинних джерел, у запаленні та як покращити біологічну активність. і посилити цілеспрямованість цих сполук. Вони потребують подальшого експериментального підтвердження.
Крім того, сигнальний шлях Nrf2 може регулювати > 600 генів [163], з яких > 200 кодують цитопротекторні білки [164], які також пов’язані із запаленням, раком, нейродегенеративними захворюваннями та іншими основними захворюваннями [165]. Все більше доказів свідчать про те, що сигнальний шлях Nrf2 дерегулюється при багатьох ракових захворюваннях, що призводить до аберантної експресії Nrf2-залежної генної батареї. Крім того, запалення відіграє важливу роль у захворюваннях, пов’язаних з окислювальним стресом, особливо при раку. Застосування кількох активаторів Nrf2 для протидії запаленню може призвести до аберантної експресії генів Nrf2 нижче по ходу, що індукує онкогенез та стійкість до хіміотерапії та/або променевої терапії. Тому для мінімізації його плейотропних ефектів можуть бути розроблені високоспецифічні активатори Nrf2. Декілька активаторів Nrf2 показали значне покращення протизапальних функцій при захворюваннях, пов’язаних з окислювальним стресом. Найкращим прикладом активатора Nrf2, схваленого FDA і широко використовуваного для лікування запальних захворювань, таких як розсіяний склероз (РС), є диметилфумарат. Tecfidera® (зареєстрована назва диметилфумарату компанією Biogen) ефективно використовується для лікування рецидивуючих форм розсіяного склерозу у великої кількості пацієнтів [152]. Однак ефективність використання активаторів Nrf2 для лікування запальних захворювань потребує подальшої перевірки, щоб уникнути шкідливих ефектів Nrf2. Тому розробка методів лікування протизапальної активності, опосередкованої Nrf2, може мати значний клінічний вплив. Постійні дослідження сигнального шляху Nrf2 у всьому світі присвячені розробці високоцільових терапевтичних засобів для контролю симптомів запалення, а також для запобігання та лікування раку, а також нейродегенеративних та інших серйозних захворювань.
На закінчення, Nrf2 визначає рівень окисного стресу в організмі людини і в кінцевому підсумку сприяє регуляції антиоксидантних і детоксикаційних ферментів і генів. Оскільки хронічне запалення, викликане підвищеним рівнем окисного стресу, асоціюється з нейродегенеративними захворюваннями, Nrf2 може відігравати істотну роль у лікуванні таких проблем зі здоров’ям, як хвороба Альцгеймера, серед інших. Обсяг нашої інформації обмежено хіропрактикою та проблемами здоров’я хребта. Щоб обговорити тему, зверніться до доктора Хіменеса або зв’яжіться з нами за адресою�915-850-0900.
Додаткова тема для обговорення: полегшення болю в коліні без операції
Біль у коліні є добре відомим симптомом, який може виникнути через різноманітні травми та/або стани коліна, у тому числі �спортивні травми. Коліно є одним із найскладніших суглобів в організмі людини, оскільки складається з перетину чотирьох кісток, чотирьох зв’язок, різних сухожиль, двох менісків і хрящів. За даними Американської академії сімейних лікарів, найпоширенішими причинами болю в коліні є підвивих колінної чашечки, тендиніт надколінка або коліно стрибуна та хвороба Осгуда-Шлаттера. Хоча біль у колінах найчастіше виникає у людей старше 60 років, біль у колінах також може виникати у дітей та підлітків. Біль у коліні можна лікувати вдома за методами RICE, однак серйозні травми коліна можуть вимагати негайної медичної допомоги, включаючи хіропрактику. �
Нейродегенеративні захворювання, такі як хвороба Альцгеймера та хвороба Паркінсона, вражають мільйони людей у всьому світі. Для лікування симптомів кількох нейродегенеративних захворювань доступні різноманітні варіанти лікування, хоча результати часто обмежені. Дослідження показали, що окислювальний стрес, викликаний як внутрішніми, так і зовнішніми факторами, може бути причиною розвитку нейродегенеративних захворювань. The транскрипційний фактор, Nrf2, було визначено, що він функціонує як головний захисний механізм проти окисного стресу. Метою статті нижче є показати наслідки Nrf2 про нейродегенеративні захворювання.
Нейродегенеративні захворювання пов’язані з накопиченням специфічних білкових агрегатів, що свідчить про тісний зв’язок між пошкодженим мозком і втратою протеостазу. Протеостаз відноситься до всіх процесів, за допомогою яких клітини контролюють кількість і згортання протеома завдяки широкій мережі, яка інтегрує регуляцію сигнальних шляхів, експресію генів і системи деградації білка. Цей огляд намагається узагальнити найбільш релевантні висновки про модуляцію транскрипції протеостазу, яку здійснює транскрипційний фактор NRF2 (ядерний фактор (еритроїдний 2)-подібний 2). NRF2 класично розглядали як головний регулятор реакції антиоксидантної клітини, хоча в даний час він стає ключовим компонентом механізму трансдукції для підтримки протеостазу. Як ми будемо обговорювати, NRF2 можна уявити як центр, який збирає аварійні сигнали, отримані від накопичення неправильно згорнутого білка, щоб побудувати скоординовану та стійку транскрипційну відповідь. Це досягається функціями NRF2, пов’язаними з контролем генів, які беруть участь у підтримці фізіології ендоплазматичної мережі, протеасоми та аутофагії.
Ядерний фактор (2)-подібний еритроїдного 2 (NRF2) - це основний білок-лейцин-блискавка, який сьогодні розглядається як головний регулятор клітинного гомеостазу. Він контролює базальну та індуковану стресом експресію понад 250 генів, які мають загальний енхансер цис-дію, який називається елементом антиоксидантної відповіді (ARE) [1], [2], [3], [4], [5]. Ці гени беруть участь у реакціях детоксикації фаз I, II і III, метаболізмі глутатіону та пероксиредоксину/тіоредоксину, виробленні НАДФН через пентозофосфатний шлях і яблучний фермент, окисленні жирних кислот, метаболізмі заліза та протеостазі [6]. Враховуючи ці широкі цитопротекторні функції, можливо, що один фармакологічний вплив на NRF2 може пом’якшити дію основних винуватців хронічних захворювань, включаючи окислювальний, запальний і протеотоксичний стрес. Роль NRF2 в модуляції антиоксидантного захисту та зняття запалення розглядалася в численних дослідженнях (оглядається в [7]). Тут ми зосередимося на його ролі в протеостазі, тобто гомеостатичному контролі синтезу білка, згортання, торгівлі та деградації. Приклади будуть наведені в контексті нейродегенеративних захворювань.
Втрата протеостазу впливає на активність NRF2 при нейродегенеративних захворюваннях
Загальною ознакою нейродегенеративних захворювань є поява аберантної агрегації деяких білків. Таким чином, неправильно згорнуті білкові агрегати ?-синуклеїну (?-SYN) виявляються в хворобі Паркінсона (PD), ?-амілоїдних (A?) бляшках і гіперфосфорильованих нейрофібрилярних клубках TAU при хворобі Альцгеймера (AD), Хантінгтіна (Htt) у Хвороба Гентінгтона (HD), супероксиддисмутаза 1 (SOD1) і TAR ДНК-зв'язуючий білок 43 (TDP-43) при боковому аміотрофічному склерозі (ALS), пріонний білок (PrP) при губчастих енцефалопатіях тощо. Білкові агрегати можуть впливати на кілька клітинні шляхи, які, у свою чергу, можуть впливати на рівень і активність NRF2.
Різні рівні регулювання жорстко контролюють активність NRF2
У фізіологічних умовах клітини демонструють низький рівень білка NRF2 через його швидкий оборот. У відповідь на різні подразники білок NRF2 накопичується, потрапляє в ядро і збільшує транскрипцію ARE-вмісних генів. Таким чином, керування рівнями білка NRF2 є ключовим моментом, який повинен інтегрувати позитивні та негативні вхідні сигнали. Як ми будемо обговорювати далі, NRF2 активується різними механізмами, що перекриваються, щоб організувати швидку та ефективну відповідь, але, з іншого боку, NRF2 може бути пригнічений, ймовірно, на другій фазі, щоб вимкнути його відповідь.
З класичної точки зору, активацію NRF2 розглядали як наслідок реакції клітини на окислювачі або електрофільні сполуки. У зв’язку з цим вирішальну роль відіграє адаптер убіквітин-Е3-лігази Kelch-подібний ECH-асоційований білок 1 (KEAP1). Молекулярні деталі будуть додатково розглянуті в Розділі 4.1. Коротше кажучи, KEAP1 діє як окислювально-відновний датчик через критичні залишки цистеїну, що призводять до убіквітинування NRF2 та протеасомної деградації. На додаток до цієї класичної модуляції, NRF2 глибоко регулюється сигнальними подіями. Дійсно, було показано, що різні кінази фосфорилюють і регулюють NRF2. Наприклад, NRF2 може фосфорилюватися мітоген-активованими протеїнкіназами (MAPK), хоча його внесок у активність NRF2 залишається неясним [8], [9], [10], [11]. PKA кіназа, а також деякі ізоферменти PKC [12], CK2 [13] або Fyn [14] фосфорилюють NRF2, змінюючи його стабільність. Попередня робота нашої групи повідомляла, що глікогенсинтаза кінсе-3? (GSK-3?) інгібує NRF2 шляхом виключення ядер і протеасомної деградації [15], [25], [26], [27], [28], [29], [30]. Молекулярні деталі будуть обговорені в Розділі 4.1. Крім того, NRF2 підпадає під інші види регулювання. Наприклад, ацетилювання NRF2 за допомогою CBP/p300 підвищує його активність [17], тоді як воно інгібується miR153, miR27a, miR142-5p і miR144 [16] або метилюванням цитозин-гуанінових островів (CG) в промоторі NRF2. [18].
Вплив білкових агрегатів на механізми регулювання NRF2
У цьому розділі ми зосередимося на тому, як накопичення неправильно згорнутого білка може вплинути на активність NRF2, надавши деякі шляхи, згадані вище, як ілюстративні приклади. По-перше, ми повинні враховувати, що накопичення білка тісно пов’язане з окислювальним пошкодженням. Дійсно, накопичення та агрегація неправильно згорнутого білка викликають аномальне виробництво активних форм кисню (АФК) з мітохондрій та інших джерел [19]. Як згадувалося вище, ROS модифікує окислювально-відновні чутливі цистеїни KEAP1, що призведе до вивільнення, стабілізації та ядерної локалізації NRF2.
Що стосується протеїнопатій, то прикладом дисрегульованих сигнальних подій, які можуть вплинути на NRF2, є гіперактивація GSK-3? в AD. GSK-3?, також відомий як TAU-кіназа, бере участь у фосфорилюванні цього білка, асоційованого з мікротрубочками, що призводить до його агрегації, утворення нейрофібрилярних клубків і переривання аксонального транспорту (оглядається в [20]). З іншого боку, ГСК-3? різко знижує рівень і активність NRF2, як зазначено вище. Незважаючи на те, що амілоїдний каскад не широко визнаний, передбачає, що токсичний A? олігомери підвищують ГСК-3? активність разом із гіперфосфорилюванням TAU та загибеллю нейронів [21], [22]. Існують різні моделі, щоб пояснити, як A? віддає перевагу GSK3-? діяльність. Наприклад, А? зв’язується з рецептором інсуліну та інгібує сигнальні шляхи PI3K та AKT, які є вирішальними для підтримки GSK-3? інактивується фосфорилюванням на його N-кінцевому залишку Ser9 [23]. З іншого боку, позаклітинний A? взаємодіє з рецепторами Frizzled, блокуючи передачу сигналів WNT [24] і знову призводить до вивільнення активного GSK-3?. Підсумовуючи, А? накопичення призводить до аномальної гіперактивації GSK-3?, таким чином погіршуючи відповідну відповідь NRF2.
Як обговорюється в наступному розділі, неправильно згорнуті білки призводять до активації PERK і MAPK, які, у свою чергу, посилюють регуляцію NRF2 [31], [8], [9], [10], [11]. Більше того, про дисрегульовану активність CBP/p300 повідомлялося при кількох протеїнопатіях [32], а також було показано глобальне зниження метилювання ДНК у мозку AD [33], що є підставою для дослідження актуальності цих висновків для регуляції NRF2.
Ми та інші спостерігали при розтинах пацієнтів із PD та AD збільшення рівня білка NRF2 та деяких його мішеней, таких як гемоксигеназа 1 (HMOX1), NADPH хіноноксидаза 1 (NQO1), p62 тощо, як за допомогою імуноблоту, так і імуногістохімією [34], [35], [36], [37], [38], [39]. Підвищення регуляції NRF2 при цих захворюваннях інтерпретується як невдала спроба хворого мозку відновити гомеостатичні значення. Однак інше дослідження показало, що NRF2 переважно локалізований в цитоплазмі нейронів гіпокампу AD, що свідчить про знижену транскрипційну активність NRF2 у мозку [40]. Цілком можливо, що невідповідність цих спостережень пов’язана зі змінами факторів, які контролюють NRF2 на прогресуючих стадіях нейродегенерації.
Три основні системи сприяють протеостазу, а саме реакція розгорнутого білка (UPR), убіквітинова протеасомна система (UPS) і аутофагія. Далі ми представляємо докази того, що NRF2 є центром, що з’єднує аварійні сигнали, активовані білковими агрегатами, з механізмом похідного білка.
NRF2 бере участь у розгорнутій білковій відповіді (UPR)
Активація NRF2 у відповідь на УПО
Окислювальне згортання білка в ER обумовлено рядом різних шляхів, найбільш консервативний з яких включає білкову дисульфід-ізомеразу (PDI) і ендоплазматичний оксидоредуктин 1 сульфгідрил оксидази (ERO1? і ERO1? у ссавців) як донора дисульфіду. Коротше кажучи, PDI каталізує утворення та розрив дисульфідних зв’язків між залишками цистеїну всередині білків, коли вони згортаються, внаслідок відновлення та окислення його власних амінокислот цистеїну. PDI переробляється під дією господарського ферменту ERO1, який знову вводить дисульфідні зв’язки в PDI [41]. Молекулярний кисень є кінцевим акцептором електронів ERO1, який генерує стехіометричні кількості перекису водню для кожного утвореного дисульфідного зв’язку [42]. Пероксидази (PRX4) і глутатіонпероксидази (GPX7 і GPX8) є ключовими ферментами для зменшення перекису водню в ER. Коли ця оксидоредуктивна система не працює належним чином, у ER відбувається аномальне накопичення неправильно згорнутих білків, і набір сигналів, названих відповіддю на розгорнутий білок (UPR), передається в цитоплазму та ядро для відновлення гомеостазу ER [43]. Ідентифіковані три мембранно-асоційовані білки для визначення стресу ER у еукаріотів: активуючий транскрипційний фактор 6 (ATF6), панкреатичний ER eIF2? кіназа (PERK, також дволанцюгова РНК-активована протеїнкіназа-подібна ER кіназа) та кіназа 1, що потребує інозитолу (IRE1). Люмінальний домен кожного датчика зв’язаний з шапероном 78 кДа, який називається глюкозо-регульованим білком (GRP78/BIP). BIP дисоціює під час стресу ER, щоб зв’язати розгорнуті білки, що призводить до активації трьох сенсорів [44].
NRF2 та його гомолог NRF1, також пов’язані з антиоксидантною відповіддю, беруть участь у трансдукції UPR до ядра. У випадку NRF1 цей білок розташований на мембрані ER і піддається ядерній транслокації після деглікозилювання або розщеплення. Потім активація UPR призводить до обробки NRF1 і ядерного накопичення отриманого фрагмента в ядерному відсіку. Проте можливість трансактивації ARE-вмісних генів цього фрагмента NRF1 все ще обговорюється [45].
Гловер-Каттер і його співробітники показали активацію ортолога NRF2 C. elegans, SKN-1, з різними стресорами ER. Підвищена експресія SKN-1 залежала від різних медіаторів UPR, включаючи ортологів хробаків IRE1 або PERK [46]. У PERK-дефіцитних клітинах порушення синтезу білка призводить до накопичення ендогенних пероксидів і подальшого апоптозу [47]. Ефектором, який використовується PERK для захисту ER від цих пероксидів, може бути NRF2, оскільки повідомлялося, що PERK фосфорилює NRF2 на Ser40, таким чином запобігаючи його деградації KEAP1 [31]. Індукція ASK1 також, ймовірно, відіграє роль у цьому шляху через TRAF2-опосередковану кіназну дію IRE1 [48]. Хоча роль MAPK у регуляції NRF2 все ще викликає суперечки, нещодавно було висловлено припущення, що шлях IRE1-TRAF2-ASK1-JNK може активувати NRF2 [49] (рис. 1). Цікаво, що в C. elegans і клітинах людини нові дані свідчать про те, що цистеїнсульфенілування кінази IRE1 в її петлі активації інгібує опосередкований IRE1 UPR і ініціює антиоксидантну відповідь p38, викликану NRF2. Дані свідчать про те, що IRE1 виконує давню функцію цитоплазматичного сторожа, який активує p38 і NRF2 [50].
Рисунок 1 Регламент NRF2 УПО. Накопичення розгорнутих або неправильно згорнутих білків всередині ендоплазматичного ретикулума може ініціювати реакцію розгорнутого білка (UPR). По-перше, шаперон BIP вивільняється з внутрішньопросвітного домену ER-сенсорів IRE1 і PERK для зв’язування розгорнутих/неправильно згорнутих білків. Це забезпечує димеризацію та транс-автофосфорилювання їхніх цитозольних доменів. Активація PERK призводить до прямого фосфорилювання NRF2 на Ser40, що призводить до транслокації NRF2 до ядра та активації цільових генів. Активація IRE1 викликає залучення TRAF2 з подальшим фосфорилюванням і активацією ASK1 і JNK. Оскільки, як повідомляється, JNK фосфорилює та активує NRF2, розумно вважати, що активація IRE1 призведе до підвищення активності NRF2.
Багато досліджень щодо індукції UPR було проведено з інгібітором глікозилювання білка тунікаміцином. NRF2, здається, має важливе значення для запобігання спричиненої тунікаміцином апоптотичної загибелі клітин [31], і його активація за цих умов зумовлена аутофагічною деградацією KEAP1 [51]. Відповідно, опосередковане shRNA приглушення експресії NRF2 в клітинах ?TC-6, лінії ?-клітин мишачої інсуліноми, значно підвищило індуковану тунікаміцином цитотоксичність і призвело до збільшення експресії проапоптотичного маркера стресу ER CHOP10. З іншого боку, активація NRF2 1,2-дитіол-3-тіоном (D3T) зменшувала цитотоксичність тунікаміцину та ослаблювала експресію CHOP10 та PERK [52]. Цікаво, що нюхові нейрони, піддані системному застосуванню тунікаміцину, збільшували NRF2 паралельно з іншими членами UPR, такими як CHOP, BIP, XBP1 [53]. Ці результати були поширені на дослідження in vivo, оскільки бічна шлуночкова інфузія тунікаміцину у щурів викликала експресію PERK і NRF2 в гіпокампі, що супроводжувалося значним когнітивним дефіцитом, збільшенням фосфорилювання TAU та відкладенням A?42 [54].
NRF2 регулює ключові гени для підтримки фізіології ER
Просвіт ER потребує великої кількості GSH з цитозолю для підтримки дисульфідної хімії. NRF2 модулює важливі ферменти метаболізму GSH в мозку, такі як транспорт цистин/глутамат, ?-глутамат цистеїнсинтетаза (?-GS), каталітичні та модуляторні субодиниці глутамат-цистеїн лігази (GCLC і GCLM), глутатіонредуктаза (GR) глутатіонпероксидази (GPX) (огляд в [55]). Відповідність NRF2 для підтримки GSH в ER підтверджується висновком, що фармакологічна або генетична активація NRF2 призводить до збільшення синтезу GSH за допомогою GCLC/GCLM, в той час як інгібування експресії цих ферментів шляхом NRF2-нокдауну спричиняє накопичення пошкоджених білки в ЕР, що призводять до активації UPR [56].
У C. elegans кілька компонентів цільових генів UPR, які регулюються SKN-1, включаючи Ire1, Xbp1 і Atf6. Хоча NRF2 посилює експресію кількох генів пероксидази (PRX) і глутатіонпероксидази (GPX) у ссавців (оглядається в [57]), лише GPX8 є справжнім ER-локалізованим ферментом, що містить сигнал пошуку KDEL [58]. Втрата GPX8 викликає активацію UPR, витік перекису водню, отриманого з ERO1?, в цитозоль і загибель клітини. Перекис водню, отриманий з ERO1? активність не може дифундувати від ER до цитозолю через узгоджену дію GPX8 і PRX4 [59]. У зв’язку з цим аналіз масиву експресії генів шляху антиоксидантного захисту з використанням РНК із тканини мишей дикого типу та нульової NRF2 показав, що експресія GPX8 була зниженою за відсутності NRF2 [60]. Відповідно до цього аналіз транскриптомів із зразків пацієнтів, які страждають на мієлопроліферативні новоутворення, поліцитемію або мієлофіброз, захворювання, також пов’язані з окислювальним стресом і хронічним запаленням низької стадії, показує нижчі рівні експресії як NRF2, так і GPX8 порівняно з контрольними суб’єктами [61]. Ще немає досліджень, які б спеціально залучали GPX8 у захист мозку людини, але аналіз транскриптомів у мишей вказує на компенсаторне збільшення GPX8 у відповідь на токсин Паркінсона MPTP [62].
Вплив NRF2 на порушення регуляції UPR при нейродегенеративних захворюваннях
Порушення функціонування ферментів PDI та хронічна активація UPR можуть згодом ініціювати або прискорювати нейродегенерацію. Нейрони, уражені захворюванням, моделі нейродегенеративних захворювань на тваринах, а також посмертні тканини людини свідчили про підвищення регуляції кількох маркерів UPR у більшості цих розладів. Зміна шляху PDI/UPR при нейродегенеративних захворюваннях була добре розглянута в [63], але слід розглянути наступні основні моменти, отримані від посмертних зразків мозку. Рівні PDI збільшуються в нейронах, що несуть клубок, і в тільцях Леві у пацієнтів з БА та PD відповідно [64], [65]. PDI і ERP57 підвищуються в спинномозковій рідині у пацієнтів з БАС і в мозку у суб’єктів CJD [66], [67], [68]. BIP, PERK, IRE1 і ATF6 підвищені у зразках пацієнтів з AD, PD або ALS [69], [70], [71], [67]. BIP, CHOP і XBP1 підвищені в посмертних зразках мозку з HD [72], [73]. Крім того, у пацієнтів з CJD в тканинах кори було виявлено підвищення ERP57, GRP94 і BIP [74]. Загалом ці дані показують, що накопичення неправильно згорнутих білків у паренхімі мозку призводить до шкідливої та хронічної активації UPR. Цікаво, що є нещодавнє дослідження, яке пов’язує активацію NRF2 PERK на початку нашої ери. У цьому дослідженні автори проаналізували, чи можуть опосередковані окисним стресом зміни в NRF2 і UPR являти собою ранні події в патогенезі AD, використовуючи клітини периферичної крові людини та модель трансгенної миші AD на різних стадіях захворювання. Підвищений окислювальний стрес і збільшення pSer40-NRF2 спостерігалися в мононуклеарних клітинах периферичної крові людини, виділених від осіб з легкими когнітивними порушеннями. Більше того, вони повідомили про порушення гомеостазу кальцію ER та підвищену регуляцію маркерів ER-стресу в цих клітинах у осіб із легкими когнітивними порушеннями та легким AD [75].
Взаємна регуляція NRF2 і убіквітинової протеасомної системи (UPS)
ДБЖ модулює рівень білка NRF2
UPS бере участь у деградації пошкоджених або неправильно згорнутих білків і контролює рівні ключових регуляторних молекул у цитозолі та ядрі. Центральним ядром цієї системи є великий багатосубодиниковий фермент, який містить протеолітичний активний комплекс під назвою 20S. Протеасома ядра 20S руйнує розгорнуті білки, але зв’язування з різними регуляторними білковими комплексами змінює її субстратну специфічність та активність. Наприклад, додавання однієї або двох регуляторних субодиниць 19S до ядра 20S утворює протеасому 26S і змінює її специфічність щодо нативних згорнутих білків [76], [77]. Протеасомна деградація потребує ковалентного зв’язування убіквітину. Кон'югація убіквітину відбувається за триступінчастим каскадним механізмом. По-перше, фермент Е1, що активує убіквітин, активує убіквітин в реакції, що вимагає АТФ. Потім один фермент Е2 (білок-переносник убіквітину або фермент, що кон’югує убіквітин) переносить активований убіквітин з Е1 на субстрат, який специфічно зв’язаний з членом сімейства убіквітин-білкових лігаз, який називається Е3. Хоча точна доля убіквітинованого білка буде залежати від природи убіквітинового ланцюга, цей процес зазвичай призводить до деградації протеасомою 26S [78].
E3-лігаза KEAP1 є найвідомішим інгібітором NRF2. Механізм регулювання KEAP1 елегантно пояснює, як рівні NRF2 пристосовуються до коливань окислювача. У базальних умовах знову синтезований NRF2 захоплюється гомодимером KEAP1, який зв’язує одну молекулу NRF2 на двох амінокислотних послідовностях з низьким (аспартат, лейцин, гліцин; DLG) і високим (глутамат, треонін, гліцин, глутамат; ETGE). Взаємодія з KEAP1 сприяє представленню NRF2 білковому комплексу CULLIN3/RBX1, що призводить до його убіквітинування та подальшої протеасомної деградації. Однак окислювально-відновна модифікація KEAP1 перешкоджає представленню NRF2 в ДБЖ, представленому CULLIN3/RBX1. В результаті нещодавно синтезований NRF2 уникає KEAP1-залежної деградації, накопичується в ядрі та активує гени, що містять ARE [79], [80], [81], [82].
Адаптер E3-лігази ?-TrCP також є гомодимером, який бере участь у сигнальних подіях, пов'язаних з фосфорилюванням NRF2 за допомогою GSK-3?. Ця кіназа фосфорилює специфічні серинові залишки NRF2 (аспартат, серин, гліцин, ізолейцин серин; DSGIS), щоб створити домен деградації, який потім розпізнається ?-TrCP і позначений для деградації протеасоми комплексом CULLIN1/RBX1. Визначення специфічних амінокислот, які фосфорилюються GSK-3? у цьому дегроні проводили комбінацію сайт-спрямованого мутагенезу домену Neh6, електрофорезу в 2D-гелі [15], [26] та мас-спектроскопії [83]. Отже, інгібування ГСК-3? високоселективними препаратами або siRNA проти ізоформ GSK-3 призводило до підвищення рівня білка NRF2. Подібні результати були знайдені з siRNA проти ізоформ 1 і 2 ?-TrCP. Стабілізація NRF2 після GSK-3? інгібування відбулося у фібробластах ембріонів миші з дефіцитом KEAP1 і в ектопічна експресованого делеційного мутанта NRF2, у якого відсутні критичні залишки ETGE для високоафінного зв’язування з KEAP1, що додатково демонструє незалежну від KEAP1 регуляцію.
У контексті нейродегенеративних захворювань ми можемо уявити модуляцію NRF2 ДБЖ двома різними способами. З одного боку, система KEAP1 буде відчувати окислювально-відновний дисбаланс, що виникає внаслідок накопичення неправильно згорнутого білка, тоді як вісь GSK-3/?-TrCP буде діяти як активний учасник сигнальної трансдукції, зміненої втратою протеостазу (рис. 2).
Малюнок 2 ДБЖ чітко контролює рівні NRF2. У гомеостатичних умовах низькі рівні NRF2 підтримуються завдяки дії адаптерів лігаз E3 KEAP1 і ?-TrCP. Ліворуч, NRF2 зв’язується з доменами Кельха гомодимеру KEAP1 через мотиви низької (DLG) і високої (ETGE) спорідненості. Через свій домен BTB KEAP1 одночасно зв’язується з комплексом CULLIN3/RBX1, забезпечуючи убіквітинування та деградацію NRF2 протеасомою 26 S. Більше того, ГСК-3? фосфорилює залишки Ser335 і Ser338 NRF2, щоб створити домен деградації (DpSGIpSL), який потім розпізнається адаптером убіквітин-лігази ?-TrCP і позначений для деградації протеасоми комплексом CULLIN3/RBX1. Праворуч, після впливу активних форм кисню або електрофілів критичні залишки Cys в KEAP1 модифікуються, через що KEAP1 не може ефективно взаємодіяти з NRF2 або CULLIN3/RBX1, а потім цей фактор транскрипції збільшує його період напіввиведення та транскрипційну активність щодо генів ARE. Сигнальні шляхи, які призводять до інгібування GSK-3?, наприклад, фосфорилювання AKT на Ser9, призводять до порушення NRF2 деградації протеасомою, накопичення та індукції цільових генів.
NRF2 збільшує активність UPS за допомогою контролю транскрипції субодиниць протеасоми
NRF2 посилює експресію кількох субодиниць протеасоми, захищаючи таким чином клітину від накопичення токсичних білків. Двадцять генів, пов’язаних із протеасомою та убіквітинацією, як видається, регулюються NRF2, згідно з широким аналізом мікрочипів із РНК печінки, який був створений за допомогою індуктора NRF2 D3T [84]. У попередньому дослідженні ті ж автори засвідчили, що експресія більшості субодиниць протеасоми 26S була збільшена втричі в печінці мишей, які отримували D3T. Рівні білка субодиниці та активність протеасоми були координовано підвищені. Однак індукції не спостерігали у мишей, де був порушений транскрипційний фактор NRF2. Активність промотора протеасомної субодиниці PSMB5 (20S) збільшувалася або при надекспресії NRF2, або при лікуванні активаторами в ембріональних фібробластах миші, а ARE були ідентифіковані в проксимальному промоторі PSMB5 [85]. Фармакологічна активація NRF2 призвела до підвищення рівня експресії репрезентативних субодиниць протеасоми (PSMA3, PSMA6, PSMB1 і PSMB5) лише у нестаріючих фібробластах людини, що містять функціональний NRF2 [86]. Активація NRF2 під час адаптації до окисного стресу призводить до високої експресії PSMB1 (20S) і PA28? субодиниць (або S11, регулятор протеасоми) [87]. Більше того, результати ембріональних стовбурових клітин людини показали, що NRF2 контролює експресію білка дозрівання протеасоми (POMP), протеасомного шаперона, який, у свою чергу, модулює проліферацію самооновлюваних людських ембріональних стовбурових клітин, диференціацію трьох зародкових листків і перепрограмування клітин [ 88]. Усі разом ці дослідження показують, що NRF2 посилює експресію ключових компонентів UPS і, отже, активно сприяє очищенню білків, які в іншому випадку були б токсичними.
Вісь NRF2-UPS при нейродегенеративних захворюваннях
Роль UPS у нейродегенеративних захворюваннях є полем інтенсивних дискусій. Початкові дослідження повідомляли про зниження активності протеасом при розтині людей у пацієнтів, уражених декількома нейродегенеративними захворюваннями. Однак інші дослідження, що застосовували підходи in vitro та in vivo, виявили незмінну або навіть підвищену активність протеасом (розглянуто в [89]). Одне з можливих пояснень цієї невідповідності полягає в тому, що рівні компонентів UPS можуть змінюватися під час прогресування захворювання та в різних областях мозку, як це було запропоновано для NRF2-цілей.
Незважаючи на цю суперечку, слід зазначити, що активізація генів протеасом, що містять ARE, посилить UPS шляхом збільшення кліренсу токсичних білків у мозку. Дійсно, абляція NRF1, також модулятора антиоксидантної реакції, в нейронних клітинах призводить до порушення активності протеасом і нейродегенерації. Експерименти з імунопреципітацією хроматину та транскрипційний аналіз продемонстрували, що PSMB6 регулюється NRF1. Крім того, профіль експресії генів призвів до ідентифікації NRF1 як ключового регулятора транскрипції генів протеасом у нейронах, що свідчить про те, що порушення в NRF1 можуть сприяти патогенезу нейродегенеративних захворювань [90]. Цікаво, що NRF1 і його довга ізоформа, яка називається TCF11, підвищують регуляцію генів протеасом, що містять ARE, після інгібування протеасоми в петлі зворотного зв’язку, щоб компенсувати знижену протеолітичну активність [91], [92].
Що стосується NRF2, то існує кореляція між зниженням рівнів NRF2, RPT6 (19 S) і PSMB5 (20 S) у середньому мозку мишей з дефіцитом DJ-1, які отримували нейротоксин паракват [93]. Крім того, природна сполука сульфорафан (SFN) дає більш надійне зображення NRF2 як важливого модулятора ДБЖ. Експерименти in vitro з клітинами нейробластоми мишей Neuro2A засвідчили посилення експресії каталітичних субодиниць протеасоми, а також її пептидазну активність у відповідь на SFN. Цей препарат захищав клітини від опосередкованої перекисом водню цитотоксичності та окислення білка залежно від функції протеасоми [94]. Крім того, Лю та його співробітники використовували мишу-репортер для моніторингу активності UPS у відповідь на SFN в мозку. Ці миші повсюдно експресують білок зеленої флуоресценції (GFP), злитий із сигналом конститутивної деградації, що сприяє його швидкому розпаду UPS (GFPu). У корі головного мозку SFN знижував рівень GFPu з паралельним збільшенням активності 5 S протеасоми, подібної до хімотрипсину (PSMB2), каспазоподібної (PSMB1) та трипсиноподібної (PSMB20). Крім того, лікування клітин Хантінгтона SFN показало, що активація NRF2 посилює деградацію mHtt і зменшує цитотоксичність mHtt [95]. Основний механізм дії SFN полягає в індукції NRF2 [96]. Специфічний внесок NRF2 слід розглядати з використанням NRF2-нульових систем у подальших дослідженнях.
Функціональний зв'язок між NRF2 і макроавтофагією
Рівні білка NRF2 модулюються адаптерним протеїном P62
Аутофагія відноситься до деградації цитозольних компонентів всередині лізосом. Цей процес використовується для очищення довгоживучих і неправильно згорнутих білків, а також пошкоджених органел. Прямий зв’язок між NRF2 та аутофагією вперше був помічений у зв’язку з адаптерним білком p62, який також називають SQSTM1 [97], [98], [99], [100], [101]. Цей білок транспортує убіквітиновані білки до механізмів деградації протеасом і лізосом і секвеструє пошкоджені білки в агрегати перед їх розпадом. P62 представляє убіквітин-асоційований (UBA) домен для зв’язування з убіквітинованими білками та ділянку, що взаємодіє з LC3 (LIR) для інтеграції з аутофагосомною мембраною через рецептор аутофагії LC3.
Хоча про опосередковану p62 індукції NRF2 та його цільових генів вперше повідомили у 2007 році [102], молекулярний механізм не був повністю зрозумілий до відкриття його взаємодії з KEAP1 [103], [98], [99], [100]. ], [101]. Komatsu та співробітники визначили ділянку взаємодії KEAP1 (KIR) у p62, яка зв'язувала KEAP1 в тій же базовій кишені на поверхні, що й NRF2, і зі спорідненістю зв'язування, подібною до мотиву ETGE в NRF2, що свідчить про конкуренцію між p62 і NRF2. Було показано, що фосфорилювання Ser351 в мотиві KIR в p62 (349-DPSTGE-354) підвищує його спорідненість до KEAP1, конкуруючи зі зв’язуванням NRF2 і дозволяючи його накопичення та транскрипційну активацію його цільових генів [98], [99]. Фактично, надекспресія p62 призвела до зниження убіквітинування NRF2 і подальшої стабілізації, а також до індукції його цільових генів [104]. Вважається, що деякі кінази беруть участь у фосфорилюванні p62. Можуть бути причетними до ссавців комплексу рапаміцину 1 (mTORC1), оскільки лікування інгібітором mTOR рапаміцином пригнічує фосфорилювання p62 та зниження регуляції KEAP1 під час лікування арсенітом. Нещодавно було продемонстровано, що TGF-α-активована кіназа 1 (TAK1) може також фосфорилувати p62, посилюючи деградацію KEAP1 і регуляцію NRF2. Автори цього дослідження припускають, що це спосіб регулювати клітинний редокстаз у стаціонарних умовах, оскільки дефіцит TAK1 підвищує АФК за відсутності будь-якого екзогенного окислювача в різних тканинах миші паралельно зі зниженням рівня білка NRF2 [105 ].
Конструкція p62 без домену UBA все ще була здатна зв’язувати KEAP1, що означає, що взаємодія не залежить від убіквітинованого KEAP1 [101]. Однак гомолог p62 у Drosophila melanogaster, названий Ref(2), не містить мотиву KIR і не взаємодіє безпосередньо з DmKEAP1, хоча він може зв’язуватися з убіквітинованим DmKEAP1 через домен UBA. Крім того, DmKEAP1 може безпосередньо взаємодіяти з Atg8 (гомологом LC3 ссавців). Дефіцит KEAP1 призводить до Atg8 та індукції аутофагії, що залежить від ортолога NRF2 CncC і не залежить від TFEB/MITF [106]. Проте зв'язок між NRF2 та аутофагією, здається, зберігається, що підкреслює його функціональне значення.
Індукція NRF2 р62 є результатом як конкуренції за зв’язування KEAP1, так і деградації KEAP1 в лізосомі. Приглушення p62 за допомогою siRNA подвоїло період напіввиведення KEAP1 паралельно зі зменшенням NRF2 та його цільових генів [101]. Відповідно, абляція експресії p62 свідчила про підвищення рівня KEAP1 порівняно з мишами дикого типу. Дуже важливо, що інгібітори протеасоми не впливали на збільшення рівнів KEAP1, але було знижено при аутофагії, що викликає голодування [107]. Фактично, KEAP1 присутній в клітинах ссавців в аутофагічних везикулах, прикрашених p62 і LC3 [99], [100], [103]. Усі ці дані свідчать про те, що KEAP1 є субстратом механізму макроавтофагії, але це питання слід проаналізувати більш детально через існування деяких суперечливих результатів. Рівні білка KEAP1 були підвищені у мишей Atg7-null, ключового ефекту макроаутофагії [107], але фармакологічне інгібування макроаутофагії за допомогою торина1, E64/пепстатину або бафіломіцину не вдалося накопичити KEAP1 [107], [100]. Загалом, ці результати свідчать про те, що підвищені рівні p62 секвеструють KEAP1 в аутофагічні вакуолі і, ймовірно, це призводить до аутофагічної деградації KEAP1, що дозволяє активувати NRF2 (рис. 3). Два різних дослідження показали, що редуктази сульфінової кислоти SESTRINS відіграють важливу роль у цьому контексті. SESTRIN 2 взаємодіє з p62, KEAP1 і RBX1 і сприяє p62-залежній деградації KEAP1 і NRF2 активації цільових генів [108]. Інше дослідження показало, що SESTRIN 2 взаємодіє з ULK1 і p62, сприяючи фосфорилюванню p62 на Ser403, що сприяє деградації білків-вантажів, включаючи KEAP1 [109].
Рисунок 3 Рівні NRF2 регулюються адаптерним білком p62. Фосфорилювання Ser 351 в мотиві KIR p62 (349-DPSTGE-354) mTORC1, TAK1 або іншими кіназами призводить до збільшення спорідненості до зв’язування з KEAP1 через схожість з мотивом ETGE в NRF2. Як наслідок, фосфорильований p62 витісняє NRF2 і зв’язує KEAP1. Мотив LIR в p62 забезпечує взаємодію з LC3 в аутофагосомальній мембрані, так що комплекс p62-KEAP1 зрештою руйнується в лізосомі. Як наслідок, NRF2 здатний накопичуватися, переміщатися в ядро і збільшувати транскрипцію ARE-вмісних генів, включаючи p62. Цей регуляторний механізм забезпечує стійку реакцію NRF2, оскільки KEAP1 має бути знову синтезований, щоб пригнічувати активність NRF2.
Модуляція генів макроаутофагії за допомогою NRF2
NRF2 регулює експресію відповідних генів для макроаутофагії, а також для UPR та UPS. Перші докази були отримані в результаті досліджень, у яких було показано, що експресія p62 індукується під впливом електрофілів, АФК та оксиду азоту [110], [111], [112]. Механізм індукції був описаний через кілька років, виявивши, що p62 містить функціональний ARE у своєму генному промоторі [99]. У недавньому дослідженні було знайдено й підтверджено декілька інших функціональних ARE за результатами біоінформаційного аналізу та аналізів ChIP. Більше того, ембріональні фібробласти миші та кортикальні нейрони мишей з нокаутом Nrf2 демонстрували знижену експресію p62, яку можна було врятувати за допомогою лентивірусу, що експресує NRF2. Аналогічно, дефіцит NRF2 знижував рівень p62 у пошкоджених нейронах гіпокампу мишей [36]. Тому було припущено, що активація NRF2 підвищує рівень p62, що призводить до деградації KEAP1 і сприяє подальшій стабілізації NRF2 у петлі позитивного зворотного зв’язку. Цей неканонічний механізм індукції NRF2 вимагає змін у експресії генів і може бути відповідною реакцією на тривалий клітинний стрес.
Було показано, що білок розпізнавання вантажу NDP52 транскрипційно регулюється NRF2. NDP52 працює подібно до p62, розпізнаючи біквітиновані білки та взаємодіючи з LC3 через домен LIR, так що вантажі розкладаються в лізосомах. У послідовності ДНК промотора Ndp52 було виявлено п'ять передбачуваних ARE. Три з них були ідентифіковані за допомогою різних мутантних конструкцій та аналізів ChIP як незамінні для NRF2-опосередкованої транскрипції Ndp52 [113]. Слід зазначити, що рівні мРНК Ndp52 були знижені в гіпокампі мишей з нокаутом Nrf2. Одна з цих послідовностей також була підтверджена в незалежному дослідженні як регульована NRF2 ARE [36].
Однак роль NRF2 в модуляції аутофагії не обмежується індукцією цих двох білків, що розпізнають вантаж. Щоб глибше зрозуміти роль NRF2 в модуляції додаткових генів, пов’язаних з аутофагією, наша група перевірила базу даних ENCODE імунопреципітації хроматину на наявність двох білків, MAFK і BACH1, які зв’язують регульовані NRF2 ARE. Використовуючи сценарій, створений з консенсусної послідовності ARE JASPAR, ми визначили кілька ймовірних ARE у багатьох генах аутофагії. Дванадцять з цих послідовностей були підтверджені як регульовані NRF2 ARE в дев'яти генах аутофагії, експресія яких була знижена у фібробластах ембріонів мишей мишей з нокаутом Nrf2, але могла бути відновлена лентивірусом, що експресує NRF2. Наше дослідження показало, що NRF2 активує експресію деяких генів, які беруть участь у різних етапах аутофагічного процесу, включаючи ініціацію аутофагії (ULK1), розпізнавання вантажу (p62 і NDP52), формування аутофагосом (ATG4D, ATG7 і GABARAPL1), елонгацію (ATG2B і ATG5). ), і кліренс аутолізосоми (ATG4D). Отже, потік аутофагії у відповідь на перекис водню був порушений, коли NRF2 був відсутній [36].
Актуальність NRF2-опосередкованої експресії генів макроаутофагії при нейродегенеративних розладах
Було показано, що дефектна аутофагія відіграє важливу роль у кількох нейродегенеративних захворюваннях [114], а абляція аутофагії призводить до нейродегенерації у мишей [115], [116]. Миші з нокаутом Atg7 виявили, що дефіцит аутофагії призводить до накопичення p62 в убіквітин-позитивних тільцях включення. KEAP1 був секвестрований в цих тілах включення, що призвело до стабілізації NRF2 та індукції цільових генів [103]. Важливо, що надмірне накопичення p62 разом з убіквітинованими білками було виявлено при нейродегенеративних захворюваннях, включаючи AD, PD та ALS [117]. Фактично, нейрони, що експресують високі рівні APP або TAU у пацієнтів з AD, також експресують p62 і ядерний NRF2, що свідчить про їхню спробу деградувати внутрішньонейронні агрегати за допомогою аутофагії [36].
Дефіцит NRF2 посилює агрегацію білка в контексті AD. Насправді, підвищені рівні фосфорильованого та нерозчинного в саркозилі TAU виявлено у мишей з нокаутом Nrf2, хоча ніякої різниці в активності кінази або фосфатази не можна було виявити порівняно з фоном дикого типу [113]. Важливо, що було продемонстровано, що NDP52 спільно локалізується з TAU в мишачих нейронах, а пряма взаємодія між фосфо-TAU і NDP52 була показана в експериментах спільної імунопреципітації як на мишах, так і на зразках AD, що вказує на його роль у деградації TAU. Цікаво, що приглушення NDP52, p62 або NRF2 в нейронах призводило до збільшення фосфо-ТАУ [113], [118]. Крім того, у гіпокампі мишей APP/PS1?E9 було виявлено збільшення внутрішньонейронних агрегатів APP, коли NRF2 був відсутній. Це корелювало зі зміненими маркерами аутофагії, включаючи збільшення співвідношення фосфо-mTOR/mTOR і phospho-p70S6k/p70S6k (вказує на пригнічення аутофагії), підвищення рівня прекатепсину D і більшої кількості мультивезикулярних тілець [119]. У мишей, які спільно експресують людський APP (V717I) і TAU (P301L), дефіцит NRF2 призвів до підвищення рівнів загального та фосфо-TAU у нерозчинній фракції та збільшення внутрішньонейронних агрегатів APP разом зі зниженим рівнем нейронів p62, NDP52, ULK1, ATG5 і GABARAPL1. Спільна локалізація між адаптерним білком p62 та APP або TAU була зменшена за відсутності NRF2 [36]. Загалом, ці результати підкреслюють важливість NRF2 в аутофагії нейронів.
Різні транскрипційні фактори діють координовано для модуляції протеостазу
В умовах стаціонарного стану протеостаз контролюється за допомогою білкових взаємодій і посттрансляційних модифікацій, що отримують швидку відповідь. Однак клітинна адаптація вимагає регуляції транскрипції генів UPR, UPS та аутофагії. Враховуючи, що нервові клітини постійно піддаються низькоякісним токсичним ураженням, включаючи окислювальний і протеотоксичний стрес, посилення протеостазу, викликане модуляцією транскрипції, може допомогти запобігти дегенерації мозку.
У випадку UPR активація кожного з трьох гілок нарешті призведе до індукції транскрипції певних генів (оглядається в [43]). Наприклад, фрагмент, отриманий з ATF6 (ATF6f) зв’язується з елементами реакції на стрес ER (ERSE) і індукує експресію кількох генів, включаючи XBPI, BIP і CHOP. Крім того, передача сигналів PERK призводить до активації транскрипційного фактора ATF4, який контролює експресію кількох пов’язаних з UPR генів та деяких інших, включаючи цільові гени NRF2 Hmox1 і p62. Нарешті, активація IRE1 призводить до генерації активного фактора транскрипції, сплайсованого XBP1 (XBP1), який контролює транскрипцію генів, що кодують білки, які беруть участь у згортанні білків.
З іншого боку, було показано, що NRF1 необхідний для експресії протеасомного гена в мозку, оскільки миші з нокаутом Nrf1 демонстрували знижену експресію генів, що кодують різні субодиниці ядра 20S, а також регуляторний комплекс 19S разом із порушенням протеасомальної функції [90]. ]. Як NRF1, так і NRF2 зв’язуються з послідовностями ARE в промоторних областях його цільових генів, що свідчить про те, що вони мають перекриваючу транскрипційну активність, хоча вони відрізняються за своїми регуляторними механізмами та клітинною локалізацією [120].
Фактори транскрипції сімейства Forkhead box O (FOXO) контролюють експресію множинних генів, пов’язаних з аутофагією. Подібно до того, що відбувається з NRF2, існує кілька рівнів регуляції активності членів FOXO, які можуть бути викликані харчовим або окислювальним стресом [121]. Нарешті, фактор транскрипції TFEB, який вважається головним регулятором лізосомного біогенезу, відіграє вирішальну роль у регуляції аутофагії в умовах харчового стресу. Таким чином, інгібування mTORC1 призводить до ядерної транслокації TFEB та індукції експресії генів аутофагії [122].
Загалом, існування різних регуляторів транскрипції цих механізмів також припускає перехресні перешкоди та частково надлишкові механізми, які можуть забезпечити протеостаз за різних обставин. Відповідно, NRF2 може відігравати відповідну роль у тканинах, які підтримують високий рівень окисного стресу. Наприклад, NRF2, спричинений окислювальним стресом, може функціонувати в умовах, багатих поживними речовинами, щоб підвищити транскрипцію аутофагії, подібно до того, що було виявлено для TFEB в умовах голодування. Більше того, мозок функціонує значною мірою в умовах, багатих поживними речовинами, представляючи NRF2 як відповідний механізм для активації аутофагії в нейронах.
Перспективний терапевтичний потенціал для NRF2 при протеїнопатіях
За останні кілька років було досягнуто значного прогресу у знанні регуляторної ролі UPR, UPS та аутофагії щодо активності NRF2, а також взаємної транскрипції, опосередкованої NRF2, компонентів цих трьох систем. Тому нові терапевтичні можливості можуть виникнути на основі використання NRF2 як важливого регулятора кліренсу білка при нейродегенеративних захворюваннях.
Однак залишається ключовим питанням, чи буде корисним чи шкідливим підвищення рівня NRF2 в мозку. Аналіз епідеміологічних даних може дати часткову відповідь, оскільки він вказує на те, що ген NFE2L2 є високополіморфним, а деякі поліморфізми окремих нуклеотидів, виявлені в його регуляторній області промотора, можуть забезпечувати діапазон «фізіологічної» мінливості в експресії генів на рівні популяції та деяких гаплотипах. були пов’язані зі зниженням ризику та/або відстроченим початком AD, PD або ALS [123]. Більше того, як обговорювали Хейз та його колеги [124], ефект NRF2 може мати U-подібну реакцію, що означає, що занадто низькі рівні NRF2 можуть призвести до втрати цитозахисту та підвищеної сприйнятливості до стресових факторів, тоді як занадто великий NRF2 може порушити гомеостатичний баланс щодо редукційний сценарій (відновлювальний стрес), який сприятиме неправильному згортанню та агрегації білка. Низькі рівні NRF2 в мозку підтверджують ідею про те, що невеликого підвищення регуляції може бути достатньо для досягнення користі при патологічних станах. Фактично, захисна роль фармакологічної NRF2-опосередкованої активації кліренсу білка була показана в різних культурах клітин нейродегенерації та моделях in vivo.
SFN є фармакологічним активатором NRF2, який, як було продемонстровано, індукує експресію генів протеасом і аутофагії [95], [36]. Цікаво, що Джо та його колеги продемонстрували, що SFN знижував рівні фосфорильованого TAU і збільшував Beclin-1 і LC3-II, припускаючи, що активація NRF2 може сприяти розпаду цього токсичного білка через аутофагію [113]. Більше того, деградація mHtt була посилена за допомогою SFN, і це було повернено за допомогою MG132, що вказує на протеасомну деградацію цього токсичного білка [95]. Повідомлялося про опосередковану аутофагією деградацію фосфо- і нерозчинного TAU з органічним флавоноїдом фізетином. Ця сполука була здатна індукувати аутофагію, одночасно сприяючи активації та транслокації ядер як TFEB, так і NRF2, а також деяких його цільових генів. Цю відповідь запобігло приглушення TFEB або NRF2 [125]. Ботт і його колеги повідомили про сприятливий вплив одночасного активатора NRF2, NRF1 і HSF1 на білкову токсичність при спінальній і бульбарній м’язовій атрофії, нейродегенеративному розладі, спричиненому розширенням повторів CAG, що кодують поліглутамін, в яких присутні білкові агрегати [126]. Потенціал активації NRF2 для лікування нейродегенеративних розладів був продемонстрований після схвалення BG-12, пероральної форми індуктора NRF2 диметилфумарату (DMF), для лікування розсіяного склерозу [127], [128]. Успіх ДМФ при аутоімунних захворюваннях із сильним запальним компонентом свідчить про те, що нейродегенеративні захворювання могли б отримати користь від репозиції цього препарату. У недавньому доклінічному дослідженні моделі ?-синуклеінопатії БП було показано, що ДМФ є нейропротекторним, частково через його індукцію аутофагії [129]. Досліджень, які повідомляють про сприятливий вплив NRF2 на нейродегенерацію, але не зосереджуються на його впливі на кліренс білка, є ще більшою кількістю (для повного огляду див. [7]). Це досить актуально, оскільки підкреслює численні ушкоджуючі процеси, які можуть бути одночасно спрямовані на один удар в NRF2, включаючи окислювальний стрес, нейрозапалення або мітохондріальну дисфункцію. Однак буде потрібна подальша робота, щоб точно визначити, чи може фармакологічна активація NRF2 бути дійсною стратегією для полегшення деградації токсичних білків у мозку.
Як пояснювалося раніше, загострюється ГСК-3? Повідомлялося про активність нейродегенеративних захворювань, і було припущено, що подальше зниження NRF2 може бути частково відповідальним за шкідливий результат. За цих патологічних умов інгібітори GSK-3 можуть також співпрацювати для підвищення рівня NRF2 і протеостазу. Про сприятливий вплив інгібіторів GSK-3 повідомлялося в різних моделях нейродегенерації, і, що більш цікаво, було показано, що репресія GSK-3 знижує рівень токсичних білків [130], [131], [132], [133]. Хоча прямих зв’язків між інгібуванням GSK-3 і регуляцією транскрипції NRF2 генів, що сприяють протеостазу, поки не спостерігалося, є розумно припустити, що зниження активності GSK-3 призведе до підвищення рівня NRF2, що в кінцевому підсумку призведе до посилення протеостаз.
Транскрипційна активність NRF2, а також здатність клітин підтримувати протеостаз зменшуються з віком, що є основним фактором ризику розвитку нейродегенеративних захворювань. Резонно вважати, що посилення NRF2 і, отже, протеостазу, принаймні сповільнить накопичення білкових агрегатів і нейродегенерацію. Дійсно, обробка старіючих фібробластів людини тритерпеноїдом 18?-гліциретинової кислоти (18?-GA) сприяла активації NRF2, що призвело до індукції протеасом і подовження тривалості життя. Це дослідження припускає, що фармакологічна активація NRF2 можлива навіть у пізньому віці [86]. Більше того, пізніше дослідження показало, що ця сполука опосередковувала SKN-1 і активацію протеасом у C.elegans з сприятливим впливом на прогресування БА у відповідних моделях нематод [134].
Враховуючи всі обставини, NRF2-опосередкована індукція генів, пов’язаних з протеостазом, здається, є корисною при різних протеїнопатіях.
Сульфорафан та його вплив на рак, смертність, старіння, мозок і поведінку, хвороби серця тощо
Ізотіоціанати є одними з найважливіших рослинних сполук, які ви можете отримати у своєму раціоні. У цьому відео я роблю для них найповнішу справу, яку коли-небудь робили. Короткий період уваги? Перейдіть до улюбленої теми, натиснувши один із моментів часу нижче. Повна хронологія нижче.
Ключові розділи:
00:01:14 – Рак і смертність
00:19:04 – Старіння
00:26:30 – Мозок і поведінка
00:38:06 – Підсумок
00:40:27 – Доза
Повний графік:
00:00:34 – Представлення сульфорафану, головна тема відео.
00:01:14 – Споживання овочів хрестоцвітних і зниження смертності від усіх причин.
00:02:12 – Ризик раку передміхурової залози.
00:02:23 – Ризик раку сечового міхура.
00:02:34 – Ризик раку легенів у курців.
00:02:48 – Ризик раку молочної залози.
00:03:13 – Гіпотетична: що робити, якщо у вас уже рак? (інтервенційний)
00:03:35 – Імовірний механізм, який керує асоціативними даними раку та смертності.
00:04:38 – Сульфорафан і рак.
00:05:32 – Докази на тваринах, що показують сильний вплив екстракту паростків брокколі на розвиток пухлин сечового міхура у щурів.
00:06:06 – Вплив прямого прийому сульфорафану у пацієнтів з раком передміхурової залози.
00:07:09 – Біоакумуляція метаболітів ізотіоціаната у фактичній тканині молочної залози.
00:08:32 – Пригнічення стовбурових клітин раку молочної залози.
00:08:53 – Урок історії: ще в Стародавньому Римі стверджували, що капустяні гриби мають оздоровчі властивості.
00:09:16 – Здатність сульфорафану посилювати виведення канцерогену (бензолу, акролеїну).
00:09:51 – NRF2 як генетичний перемикач через елементи антиоксидантної реакції.
00:10:10 – Як активація NRF2 посилює виведення канцерогену через глутатіон-S-кон'югати.
00:10:34 – Брюссельська капуста підвищує глутатіон-S-трансферазу і зменшує пошкодження ДНК.
00:11:20 – Напій з проростків брокколі збільшує виведення бензолу на 61%.
00:15:45 – Споживання хрестоцвітних овочів і смертність від серцево-судинних захворювань.
00:16:55 – порошок паростків брокколі покращує рівень ліпідів у крові та загальний ризик серцевих захворювань у діабетиків 2 типу.
00:19:04 – Початок секції старіння.
00:19:21 – Дієта, збагачена сульфорафаном, збільшує тривалість життя жуків від 15 до 30% (за певних умов).
00:20:34 – Важливість слабкого запалення для довголіття.
00:22:05 – Овочі хрестоцвітних і порошок паростків брокколі, здається, зменшують широкий спектр запальних маркерів у людей.
00:23:40 – Підсумок у середині відео: розділи про рак, старіння
00:24:14 – Дослідження на мишах показують, що сульфорафан може покращити адаптивну імунну функцію в літньому віці.
00:25:18 – Сульфорафан покращив ріст волосся у мишачої моделі облисіння. Зображення на 00:26:10.
00:26:30 – Початок розділу «Мозок і поведінка».
00:27:18 – Вплив екстракту паростків брокколі на аутизм.
00:27:48 – Вплив глюкорафаніну на шизофренію.
00:28:17 – Початок обговорення депресії (правдоподібний механізм та дослідження).
00:31:21 – Дослідження на мишах з використанням 10 різних моделей депресії, викликаної стресом, показало, що сульфорафан так само ефективний, як і флуоксетин (прозак).
00:32:00 – Дослідження показує, що пряме вживання глюкорафаніну мишами так само ефективне для запобігання депресії через модель стресу соціальної поразки.
00:33:01 – Початок відділу нейродегенерації.
00:33:30 – Сульфорафан і хвороба Альцгеймера.
00:33:44 – Сульфорафан і хвороба Паркінсона.
00:33:51 – Сульфорафан і хвороба Хантінгтона.
00:34:13 – Сульфорафан збільшує кількість білків теплового шоку.
00:34:43 – Початок секції черепно-мозкової травми.
00:35:01 – Сульфорафан, введений відразу після ЧМТ, покращує пам’ять (дослідження на мишах).
00:35:55 – Сульфорафан і нейрональна пластичність.
00:36:32 – Сульфорафан покращує навчання на моделі діабету ІІ типу у мишей.
00:37:19 – Сульфорафанова і м’язова дистрофія Дюшенна.
00:37:44 – Інгібування міостатину в клітинах-супутниках м’язів (in vitro).
00:38:06 – Пізнє відео: смертність і рак, пошкодження ДНК, окислювальний стрес і запалення, виділення бензолу, серцево-судинні захворювання, діабет ІІ типу, вплив на мозок (депресія, аутизм, шизофренія, нейродегенерація), шлях NRF2.
00:40:27 – Думки щодо визначення дози паростків брокколі або сульфорафану.
00:41:01 – Анекдоти про проростання в домашніх умовах.
00:43:14 – Про температуру приготування та активність сульфорафану.
00:43:45 – Перетворення сульфорафану з глюкорафаніну кишковими бактеріями.
00:44:24 – Добавки працюють краще в поєднанні з активною мирозиназою з овочів.
00:44:56 – Техніка приготування та овочі хрестоцвітних.
00:46:06 – Ізотіоціанати як зоб.
Фактор 2, пов’язаний з ядерним фактором еритроїдного походження (NF-E2), інакше відомий як Nrf2, є фактором транскрипції, який регулює експресію різноманітних антиоксидантних і детоксикаційних ферментів. Дослідження також продемонстрували його роль у контролі окисного стресу. Більшість нейродегенеративних захворювань, таких як хвороба Альцгеймера і хвороба Паркінсона, характеризуються окислювальним стресом і хронічним запаленням, які є загальними мішенями для Підходи до лікування Nrf2. Д-р Алекс Хіменес, округ Колумбія, CCST Insight
Заключні зауваження
Фактор транскрипції NRF2 організовує протеостатичну відповідь, відчуваючи та модулюючи зміни в UPR, UPS та аутофагії (рис. 4). Отже, було показано, що відсутність NRF2 посилює протеїнопатію, що свідчить про те, що NRF2 необхідний для оптимального кліренсу білка. Разом ми можемо припустити, що NRF2 може бути цікавою терапевтичною мішенню для протеїнопатій.
Малюнок 4 NRF2 як концентратор, що з'єднує протеотоксичні сигнали екстреної допомоги з захисною реакцією транскрипції. Накопичення розгорнутих/неправильно згорнутих білків призведе до активації реакції розгорнутого білка (UPR) в ER. Активація PERK або MAPK може призвести до індукції транскрипції резидентного ER Gpx8 і кількох ферментів, що регулюють рівень GSH, що є критичним для забезпечення правильного згортання білка. Білкові агрегати пригнічують активність протеасом (UPS), ймовірно, уникаючи деградації NRF2. Було показано, що NRF2 специфічно модулює транскрипцію генів Psma3, Psma6, Psmb1, Psmb5 і Pomp. Кілька інших субодиниць були підвищені в залежності від NRF2 у відповідь на D3T, що, ймовірно, розширило список субодиниць протеасоми, які регулюються NRF2. Аутофагія є основним шляхом деградації білкових агрегатів. Аутофагія також регулює NRF2, з'єднуючи цей шлях деградації з індукцією транскрипції NRF2 p62, Ndp52, Ulk1, Atg2b, Atg4c, Atg5, Atg7 і Gabarapl1.
Згідно зі статтею вище, хоча симптоми нейродегенеративних захворювань можна лікувати за допомогою різних варіантів лікування, дослідження показали, що активація Nrf2 може бути корисним підходом до лікування. Тому що Активатори Nrf2 націлені на широкі механізми захворювання, всі нейродегенеративні захворювання можуть отримати користь від використання фактора транскрипції Nrf2. Висновки Nrf2 зробили революцію в лікуванні нейродегенеративних захворювань. Обсяг нашої інформації обмежено хіропрактикою та проблемами здоров’я хребта. Щоб обговорити тему, зверніться до доктора Хіменеса або зв’яжіться з нами за адресою�915-850-0900.
Додаткова тема для обговорення: полегшення болю в коліні без операції
Біль у коліні є добре відомим симптомом, який може виникнути через різноманітні травми та/або стани коліна, у тому числі �спортивні травми. Коліно є одним із найскладніших суглобів в організмі людини, оскільки складається з перетину чотирьох кісток, чотирьох зв’язок, різних сухожиль, двох менісків і хрящів. За даними Американської академії сімейних лікарів, найпоширенішими причинами болю в коліні є підвивих колінної чашечки, тендиніт надколінка або коліно стрибуна та хвороба Осгуда-Шлаттера. Хоча біль у колінах найчастіше виникає у людей старше 60 років, біль у колінах також може виникати у дітей та підлітків. Біль у коліні можна лікувати вдома за методами RICE, однак серйозні травми коліна можуть вимагати негайної медичної допомоги, включаючи хіропрактику. �
Окислювальний стрес описується як пошкодження клітин, спричинене вільними радикалами або нестабільними молекулами, що в кінцевому підсумку може вплинути на здорову функцію. Людський організм створює вільні радикали для нейтралізації бактерій і вірусів, однак зовнішні фактори, такі як кисень, забруднення та радіація, часто також можуть виробляти вільні радикали. Окислювальний стрес пов'язаний з численними проблемами зі здоров'ям.
Окислювальний стрес та інші стресори включають внутрішні захисні механізми, які можуть допомогти регулювати антиоксидантну реакцію людського організму. Nrf2 – це білок, який відчуває рівень окисного стресу і дає можливість клітинам захищатися від внутрішніх і зовнішніх факторів. Також було продемонстровано, що Nrf2 допомагає регулювати гени, які беруть участь у виробництві антиоксидантних ферментів і генів стресової реакції. Мета статті нижче – пояснити ефект Nrf2 при раку.
абстрактний
Шлях Keap1-Nrf2 є основним регулятором цитопротекторних реакцій на окислювальний та електрофільний стрес. Хоча клітинні сигнальні шляхи, що ініціюються транскрипційним фактором Nrf2, запобігають ініціації та прогресуванню раку в нормальних і передракових тканинах, у повністю злоякісних клітинах активність Nrf2 забезпечує перевагу росту за рахунок збільшення хіміорезистентності раку та посилення росту пухлинних клітин. У цьому графічному огляді ми надаємо огляд шляху Keap1-Nrf2 та його порушення в ракових клітинах. Ми також коротко підсумовуємо наслідки конститутивної активації Nrf2 в ракових клітинах і як це можна використати в генній терапії раку.
Ключові слова:Nrf2, Keap1, Рак, Елемент антиоксидантної реакції, Генна терапія
Вступ
Шлях Keap1-Nrf2 є основним регулятором цитопротекторних реакцій на ендогенні та екзогенні стреси, спричинені активними формами кисню (АФК) та електрофілами [1]. Ключовими сигнальними білками в цьому шляху є транскрипційний фактор Nrf2 (фактор 2, пов'язаний з ядерним фактором еритроїду 2), який зв'язується разом з невеликими білками Maf з елементом антиоксидантної відповіді (ARE) в регуляторних областях цільових генів, і Keap1 (Kelch ECH асоціюючий білок 1), білок-репресор, який зв’язується з Nrf2 і сприяє його деградації шляхом протеасомного шляху убіквітину (рис. 1). Keap1 — це дуже багатий на цистеїн білок, Keap1 миші має загалом 25, а людський — 27 залишків цистеїну, більшість з яких може бути модифікована in vitro різними окислювачами та електрофілами [2]. Було показано, що три з цих залишків, C151, C273 і C288, відіграють функціональну роль, змінюючи конформацію Keap1, що призводить до ядерної транслокації Nrf2 і подальшої експресії цільового гена [3] (рис. 1). Точний механізм, завдяки якому модифікації цистеїну в Keap1 призводять до активації Nrf2, невідомий, але двома переважаючими, але не взаємовиключними моделями, є (1) модель «шарнір та засувка», в якій модифікації Keap1 в тіолових залишках, що знаходяться в IVR Keap1. порушують взаємодію з Nrf2, викликаючи зміщення залишків лізину всередині Nrf2, який більше не може бути поліубіквітинілований, і (2) модель, в якій модифікація тіолу викликає дисоціацію Cul3 від Keap1 [3]. В обох моделях модифікований індуктором і зв’язаний з Nrf2 Keap1 інактивується, і, отже, новосинтезовані білки Nrf2 обходять Keap1 і транслокуються в ядро, зв’язуються з ARE і стимулюють експресію цільових генів Nrf2, таких як NAD(P)H. хіноноксидоредуктаза 1 (NQO1), гемоксигеназа 1 (HMOX1), глутамат-цистеїн лігаза (GCL) і глутатіон S трансферази (GSTs) (рис. 2). На додаток до модифікацій тіолів Keap1, що призводять до індукції цільового гена Nrf2, білки, такі як p21 і p62, можуть зв’язуватися з Nrf2 або Keap1, тим самим порушуючи взаємодію між Nrf2 і Keap1 [1], [3] (рис. 3).
Рис. 1. Структури Nrf2 і Keap1 і цистеїновий код. (A) Nrf2 складається з 589 амінокислот і має шість еволюційно висококонсервативних доменів Neh1-6. Neh1 містить мотив bZip, структуру основної області – лейцинової блискавки (L-Zip), де основна область відповідає за розпізнавання ДНК, а L-Zip опосередковує димеризацію невеликими білками Maf. Neh6 функціонує як дегрон для опосередкування деградації Nrf2 в ядрі. Neh4 і 5 є трансактиваційними доменами. Neh2 містить мотиви ETGE та DLG, які необхідні для взаємодії з Keap1, і гідрофільну область залишків лізину (7 K), які є незамінними для Keap1-залежного поліубіквітинування та деградації Nrf2. (B) Keap1 складається з 624 амінокислотних залишків і має п’ять доменів. Два мотиви взаємодії білка та білка, домен BTB і домен Kelch, розділені проміжною областю (IVR). Домен BTB разом з N-кінцевою частиною IVR опосередковує гомодимеризацію Keap1 і зв’язування з Cullin3 (Cul3). Домен Кельха і С-кінцева область опосередковують взаємодію з Neh2. (C) Nrf2 взаємодіє з двома молекулами Keap1 через мотиви Neh2 ETGE та DLG. І ETGE, і DLG зв’язуються з подібними ділянками на нижній поверхні мотиву Keap1 Kelch. (D) Keap1 багатий залишками цистеїну, з 27 цистеїнами в білку людини. Деякі з цих цистеїнів розташовані поблизу основних залишків і тому є чудовими мішенями для електрофілів і окисників. Схема модифікації залишків цистеїну електрофілами відома як цистеїновий код. Гіпотеза цистеїнового коду передбачає, що структурно різні агенти, що активують Nrf2, впливають на різні цистеїни Keap1. Модифікації цистеїну призводять до конформаційних змін у Keap1, порушуючи взаємодію між доменами Nrf2 DLG і Keap1 Kelch, таким чином інгібуючи поліубіквітинування Nrf2. Показано функціональне значення Cys151, Cys273 і Cys288, оскільки Cys273 і Cys288 необхідні для придушення Nrf2 і Cys151 для активації Nrf2 індукторами [1], [3].
Рис. 2. Сигнальний шлях Nrf2-Keap1. (A і B) у базальних умовах дві молекули Keap1 зв’язуються з Nrf2, а Nrf2 поліубіквітильується лігазним комплексом E3 на основі Cul3. Ця поліубіквітилація призводить до швидкої деградації Nrf2 протеасомою. Невелика частина Nrf2 виривається з гальмівного комплексу і накопичується в ядрі, щоб опосередковувати базальну експресію ARE-залежного гена, тим самим підтримуючи клітинний гомеостаз. (C) В умовах стресу індуктори модифікують цистеїни Keap1, що призводить до інгібування убіквітилювання Nrf2 шляхом дисоціації гальмівного комплексу. (D) Відповідно до моделі шарніра та засувки, модифікація специфічних залишків цистеїну Keap1 призводить до конформаційних змін у Keap1, що призводить до відриву мотиву Nrf2 DLG від Keap1. Убіквітинування Nrf2 порушується, але зв'язування з мотивом ETGE залишається. (E) У моделі дисоціації Keap1-Cul3 зв'язування Keap1 і Cul3 порушується у відповідь на електрофіли, що призводить до втечі Nrf2 із системи убіквітування. В обох запропонованих моделях модифікований індуктором і зв’язаний з Nrf2 Keap1 інактивується, і, отже, новосинтезовані білки Nrf2 обходять Keap1 і транслокуються в ядро, зв’язуються з елементом антиоксидантної відповіді (ARE) і стимулюють експресію мішені Nrf2. гени, такі як NQO1, HMOX1, GCL та GSTs [1], [3].
Рис. 3. Механізми конститутивного ядерного накопичення Nrf2 при раку. (A) Соматичні мутації в Nrf2 або Keap1 порушують взаємодію цих двох білків. У Nrf2 мутації впливають на мотиви ETGE і DLG, але в Keap1 мутації розподілені більш рівномірно. Крім того, активація онкогену, такого як KrasG12D[5], або порушення роботи пухлинних супресорів, таких як PTEN [11], може призвести до індукції транскрипції Nrf2 і збільшення ядерного Nrf2. (B) Гіперметилювання промотору Keap1 при раку легенів і простати призводить до зниження експресії мРНК Keap1, що збільшує ядерне накопичення Nrf2 [6], [7]. (C) При сімейній папілярній карциномі нирки втрата активності ферменту фумаратгідратази призводить до накопичення фумарату і подальшого сукцинації залишків цистеїну Keap1 (2SC). Ця посттрансляційна модифікація призводить до порушення взаємодії Keap1-Nrf2 і ядерного накопичення Nrf2 [8], [9]. (D) Накопичення білків-розривників, таких як p62 і p21, може порушити зв'язування Nrf2-Keap1 і призвести до збільшення ядерного Nrf2. p62 зв'язується з Keap1, перекриваючи кишеню зв'язування для Nrf2, а p21 безпосередньо взаємодіє з мотивами DLG і ETGE Nrf2, таким чином конкуруючи з Keap1 [10].
Механізми активації та порушення регуляції Nrf2 при раку
Хоча цитозахист, що забезпечується активацією Nrf2, важливий для хіміопрофілактики раку в нормальних і передракових тканинах, у повністю злоякісних клітинах активність Nrf2 забезпечує перевагу росту за рахунок збільшення хіміорезистентності раку та посилення росту пухлинних клітин [4]. Було описано кілька механізмів, за допомогою яких сигнальний шлях Nrf2 конститутивно активується при різних видах раку: (1) соматичні мутації в Keap1 або Keap1 зв’язуючому домені Nrf2, що порушують їх взаємодію; (2) епігенетичне приглушення експресії Keap1, що призводить до дефектної репресії Nrf2; (3) накопичення білків-деструкторів, таких як p62, що призводить до дисоціації комплексу Keap1-Nrf2; (4) індукція транскрипції Nrf2 онкогенними K-Ras, B-Raf і c-Myc; і (5) посттрансляційна модифікація цистеїнів Keap1 шляхом сукцинилування, що відбувається при сімейній папілярній карциномі нирки через втрату активності ферменту фумарат гідратази [3], [4], [5], [6], [7], [ 8], [9], [10] (рис. 3). Конститутивно багатий білок Nrf2 викликає посилення експресії генів, які беруть участь у метаболізмі ліків, тим самим підвищуючи стійкість до хіміотерапевтичних препаратів та променевої терапії. Крім того, високий рівень білка Nrf2 пов’язаний з поганим прогнозом при раку [4]. Надактивний Nrf2 також впливає на проліферацію клітин, спрямовуючи глюкозу та глутамін на анаболічні шляхи, посилюючи синтез пуринів і впливаючи на пентозофосфатний шлях, щоб сприяти проліферації клітин [11] (рис. 4).
Рис. 4. Подвійна роль Nrf2 в онкогенезі. У фізіологічних умовах низьких рівнів ядерного Nrf2 достатньо для підтримки клітинного гомеостазу. Nrf2 пригнічує ініціацію пухлини та метастазування раку, усуваючи канцерогени, АФК та інші агенти, що ушкоджують ДНК. Під час пухлиногенезу накопичення пошкодження ДНК призводить до конститутивної гіперактивності Nrf2, яка допомагає автономним злоякісним клітинам витримувати високі рівні ендогенних АФК та уникати апоптозу. Постійно підвищені рівні ядерного Nrf2 активують метаболічні гени на додаток до цитопротекторних генів, що сприяють метаболічному перепрограмуванню та посиленню проліферації клітин. Рак з високим рівнем Nrf2 асоціюється з поганим прогнозом через радіо- та хіміорезистентність та агресивну проліферацію ракових клітин. Таким чином, активність шляху Nrf2 є захисною на ранніх стадіях пухлиногенезу, але шкідливою на пізніх стадіях. Таким чином, для профілактики раку підвищення активності Nrf2 залишається важливим підходом, тоді як для лікування раку інгібування Nrf2 є бажаним [4], [11].
Враховуючи, що висока активність Nrf2 зазвичай спостерігається в ракових клітинах з несприятливими результатами, існує потреба в терапії для інгібування Nrf2. На жаль, через структурну схожість з деякими іншими членами сімейства bZip, розробка специфічних інгібіторів Nrf2 є складним завданням, і на сьогоднішній день опубліковано лише кілька досліджень інгібування Nrf2. Відбираючи натуральні продукти, Ren et al. [12] визначили протипухлинну сполуку брусатол як інгібітор Nrf2, що підвищує хіміотерапевтичну ефективність цисплатину. Крім того, інгібітори PI3K [11], [13] та siRNA Nrf2 [14] були використані для інгібування Nrf2 в ракових клітинах. Нещодавно ми використали альтернативний підхід, відомий як генна терапія раку, щоб націлити ракові клітини з високим рівнем Nrf2. Лентивірусні вектори, керовані Nrf2 [15], що містять тимідинкіназу (TK), переносяться в ракові клітини з високою активністю ARE, і клітини обробляються пролікарським препаратом ганцикловіром (GCV). GCV метаболізується до GCV-монофосфату, який далі фосфорилюється клітинними кіназами в токсичну трифосфатну форму [16] (рис. 5). Це призводить до ефективного знищення не тільки пухлинних клітин, що містять ТЗ, але й сусідніх клітин завдяки ефекту стороннього спостерігача [17]. Регульована ARE генна терапія TK/GCV може бути додатково покращена шляхом поєднання хіміотерапевтичного агента раку доксорубіцину з лікуванням [16], що підтверджує думку про те, що цей підхід може бути корисним у поєднанні з традиційною терапією.
Рис. 5. Генна терапія суїциду. Конститутивне накопичення ядер Nrf2 в ракових клітинах можна використати за допомогою Nrf2-керованого вірусного вектора для генної терапії раку [16]. У цьому підході лентивірусний вектор (LV), що експресує тимідинкіназу (TK) під мінімальним промотором SV40 з чотирма ARE, трансдукується в клітини аденокарциноми легенів. Високі рівні ядерного Nrf2 призводять до надійної експресії TK через зв’язування Nrf2. Потім клітини обробляють пролікарським препаратом, ганцикловіром (GCV), який фосфорилюється ТЗ. Трифосфорильований GCV порушує синтез ДНК і призводить до ефективного знищення не тільки пухлинних клітин, що містять ТЗ, а й сусідніх клітин завдяки ефекту стороннього спостерігача.
Nrf2 є головним регулятором, який запускає виробництво потужних антиоксидантів в організмі людини, які допомагають усунути окислювальний стрес. Різні антиоксидантні ферменти, такі як супероксиддисмутаза або СОД, глутатіон і каталаза, також активуються через шлях Nrf2. Крім того, деякі фітохімічні речовини, такі як куркума, ашваганда, бакопа, зелений чай і розторопша, активують Nrf2. Дослідження показали, що Активація Nrf2 може природним чином посилити захист клітин і відновити баланс людського тіла.
Д-р Алекс Хіменес, округ Колумбія, CCST Insight
Сульфорафан та його вплив на рак, смертність, старіння, мозок і поведінку, хвороби серця тощо
Ізотіоціанати є одними з найважливіших рослинних сполук, які ви можете отримати у своєму раціоні. У цьому відео я роблю для них найповнішу справу, яку коли-небудь робили. Короткий період уваги? Перейдіть до улюбленої теми, натиснувши один із моментів часу нижче. Повна хронологія нижче.
Ключові розділи:
00:01:14 – Рак і смертність
00:19:04 – Старіння
00:26:30 – Мозок і поведінка
00:38:06 – Підсумок
00:40:27 – Доза
Повний графік:
00:00:34 – Представлення сульфорафану, головна тема відео.
00:01:14 – Споживання овочів хрестоцвітних і зниження смертності від усіх причин.
00:02:12 – Ризик раку передміхурової залози.
00:02:23 – Ризик раку сечового міхура.
00:02:34 – Ризик раку легенів у курців.
00:02:48 – Ризик раку молочної залози.
00:03:13 – Гіпотетична: що робити, якщо у вас уже рак? (інтервенційний)
00:03:35 – Імовірний механізм, який керує асоціативними даними раку та смертності.
00:04:38 – Сульфорафан і рак.
00:05:32 – Докази на тваринах, що показують сильний вплив екстракту паростків брокколі на розвиток пухлин сечового міхура у щурів.
00:06:06 – Вплив прямого прийому сульфорафану у пацієнтів з раком передміхурової залози.
00:07:09 – Біоакумуляція метаболітів ізотіоціаната у фактичній тканині молочної залози.
00:08:32 – Пригнічення стовбурових клітин раку молочної залози.
00:08:53 – Урок історії: ще в Стародавньому Римі стверджували, що капустяні гриби мають оздоровчі властивості.
00:09:16 – Здатність сульфорафану посилювати виведення канцерогену (бензолу, акролеїну).
00:09:51 – NRF2 як генетичний перемикач через елементи антиоксидантної реакції.
00:10:10 – Як активація NRF2 посилює виведення канцерогену через глутатіон-S-кон'югати.
00:10:34 – Брюссельська капуста підвищує глутатіон-S-трансферазу і зменшує пошкодження ДНК.
00:11:20 – Напій з проростків брокколі збільшує виведення бензолу на 61%.
00:15:45 – Споживання хрестоцвітних овочів і смертність від серцево-судинних захворювань.
00:16:55 – порошок паростків брокколі покращує рівень ліпідів у крові та загальний ризик серцевих захворювань у діабетиків 2 типу.
00:19:04 – Початок секції старіння.
00:19:21 – Дієта, збагачена сульфорафаном, збільшує тривалість життя жуків від 15 до 30% (за певних умов).
00:20:34 – Важливість слабкого запалення для довголіття.
00:22:05 – Овочі хрестоцвітних і порошок паростків брокколі, здається, зменшують широкий спектр запальних маркерів у людей.
00:23:40 – Підсумок у середині відео: розділи про рак, старіння
00:24:14 – Дослідження на мишах показують, що сульфорафан може покращити адаптивну імунну функцію в літньому віці.
00:25:18 – Сульфорафан покращив ріст волосся у мишачої моделі облисіння. Зображення на 00:26:10.
00:26:30 – Початок розділу «Мозок і поведінка».
00:27:18 – Вплив екстракту паростків брокколі на аутизм.
00:27:48 – Вплив глюкорафаніну на шизофренію.
00:28:17 – Початок обговорення депресії (правдоподібний механізм та дослідження).
00:31:21 – Дослідження на мишах з використанням 10 різних моделей депресії, викликаної стресом, показало, що сульфорафан так само ефективний, як і флуоксетин (прозак).
00:32:00 – Дослідження показує, що пряме вживання глюкорафаніну мишами так само ефективне для запобігання депресії через модель стресу соціальної поразки.
00:33:01 – Початок відділу нейродегенерації.
00:33:30 – Сульфорафан і хвороба Альцгеймера.
00:33:44 – Сульфорафан і хвороба Паркінсона.
00:33:51 – Сульфорафан і хвороба Хантінгтона.
00:34:13 – Сульфорафан збільшує кількість білків теплового шоку.
00:34:43 – Початок секції черепно-мозкової травми.
00:35:01 – Сульфорафан, введений відразу після ЧМТ, покращує пам’ять (дослідження на мишах).
00:35:55 – Сульфорафан і нейрональна пластичність.
00:36:32 – Сульфорафан покращує навчання на моделі діабету ІІ типу у мишей.
00:37:19 – Сульфорафанова і м’язова дистрофія Дюшенна.
00:37:44 – Інгібування міостатину в клітинах-супутниках м’язів (in vitro).
00:38:06 – Пізнє відео: смертність і рак, пошкодження ДНК, окислювальний стрес і запалення, виділення бензолу, серцево-судинні захворювання, діабет ІІ типу, вплив на мозок (депресія, аутизм, шизофренія, нейродегенерація), шлях NRF2.
00:40:27 – Думки щодо визначення дози паростків брокколі або сульфорафану.
00:41:01 – Анекдоти про проростання в домашніх умовах.
00:43:14 – Про температуру приготування та активність сульфорафану.
00:43:45 – Перетворення сульфорафану з глюкорафаніну кишковими бактеріями.
00:44:24 – Добавки працюють краще в поєднанні з активною мирозиназою з овочів.
00:44:56 – Техніка приготування та овочі хрестоцвітних.
00:46:06 – Ізотіоціанати як зоб.
Подяки
Ця робота була підтримана Академією Фінляндії, Фондом Сігріда Юселіуса та фінськими онкологічними організаціями.
Як висновок, ядерний фактор (2), подібний до еритроїду, також відомий як NFE2L2 або Nrf2, є білком, який збільшує вироблення антиоксидантів, які захищають організм людини від окисного стресу. Як описано вище, стимуляція шляху Nrf2 досліджується для лікування захворювань, спричинених окислювальним стресом, включаючи рак. Обсяг нашої інформації обмежено хіропрактикою та проблемами здоров’я хребта. Щоб обговорити тему, зверніться до доктора Хіменеса або зв’яжіться з нами за адресою�915-850-0900.
Додаткова тема для обговорення: полегшення болю в коліні без операції
Біль у коліні є добре відомим симптомом, який може виникнути через різноманітні травми та/або стани коліна, у тому числі �спортивні травми. Коліно є одним із найскладніших суглобів в організмі людини, оскільки складається з перетину чотирьох кісток, чотирьох зв’язок, різних сухожиль, двох менісків і хрящів. За даними Американської академії сімейних лікарів, найпоширенішими причинами болю в коліні є підвивих колінної чашечки, тендиніт надколінка або коліно стрибуна та хвороба Осгуда-Шлаттера. Хоча біль у колінах найчастіше виникає у людей старше 60 років, біль у колінах також може виникати у дітей та підлітків. Біль у коліні можна лікувати вдома за методами RICE, однак серйозні травми коліна можуть вимагати негайної медичної допомоги, включаючи хіропрактику.
ДНК підтримує приблизно 20,000 XNUMX генів, кожен з яких має програму для створення білка або ферменту, необхідного для здорового способу життя. Кожен із цих шаблонів повинен постійно регулюватися свого роду «промотором», який точно керує, скільки кожної речовини та/або хімічної речовини утворюється та за яких умов вони також розвиватимуться.
Підключаючись до певного типу промоторних областей, подібних до перемикача, відомих як елемент антиоксидантної реакції, або ARE, Nrf2 фактор�підтримує швидкість створення сотень різних генів, які дозволяють клітинам виживати в стресових умовах. Потім ці гени генерують добір антиоксидантних ферментів, які розвивають захисну мережу, нейтралізуючи окислювачі та очищаючи токсичні побічні продукти, що залишаються при їх виробництві, а також допомагають відновити пошкодження, які вони завдали.
Що таке окислювальний стрес?
Кілька окислювачів, таких як супероксидний радикал, або O2-, і перекис водню, або H2O2, були створені завдяки практиці спалювання речовин та/або хімічних речовин, які підтримують організм людини. Організм людини має антиоксидантні ферменти, які нейтралізують і детоксикують реактивні продукти та напої, які ми споживаємо. Nrf2 модулює їх виробництво, щоб підтримувати рівновагу, і підкреслює попит на всі ці ферменти. Цей баланс може бути порушений кількома факторами, зокрема віком.
З віком людський організм виробляє менше Nrf2, і ця тонка рівновага може поступово почати переходити в сторону окислення, стан, який називають окислювальним стресом. Хвороба також може викликати надлишкове виробництво окислювачів. Інфекції, алергії та аутоімунні розлади можуть додатково викликати у наших імунних клітинах утворення реактивних окислювачів, таких як O2-. , H2O2, OH і HOCl, де здорові клітини пошкоджуються і реагують на запалення. Захворювання, пов’язані зі старінням, включаючи серцеві напади, інсульт, рак і нейродегенеративні стани, такі як хвороба Альцгеймера, також збільшують вироблення окислювачів, викликаючи стрес і реакцію на запалення.
Що таке активатори Nrf2?
Білок Nrf2, також званий фактором транскрипції через те, як він може підтримувати та контролювати ферменти та гени, є секретним елементом послідовності біохімічних реакцій всередині клітини, який реагує на зміни когнітивної рівноваги, а також на окислювальний баланс. Чутливі елементи цього шляху модифікують і вивільняють Nrf2, запускаючи його, щоб він міг поширитися в ядро клітини до ДНК. Nrf2 може в якості альтернативи вмикати або вимкнути гени та ферменти, які він підтримує для захисту клітини.
На щастя, різноманітні речовини, які є активаторами Nrf2, утворюються внаслідок споживання деяких рослин та екстрактів, які використовувалися століття тому в традиційних засобах китайської та індіанської медицини. Ці фітохімічні речовини, здається, настільки ж потужні з меншою кількістю побічних ефектів, як і фармацевтичні продукти, що активують Nrf2, які використовуються сьогодні.
Фактор, пов’язаний з ядерним фактором еритроїду 2, більш відомий як Nrf2, є фактором транскрипції, який захищає клітину, регулюючи гени, ферменти та антиоксидантні реакції. Фактори транскрипції — це тип білка, який приєднується до ДНК, щоб сприяти створенню специфічних речовин і хімічних речовин, включаючи глутатіон-S-трансферазу або GST. Активація Nrf2 викликає вироблення активних білків, які виявляють потужну антиоксидантну здатність, щоб допомогти зменшити окислювальний стрес.
Д-р Алекс Хіменес, округ Колумбія, CCST Insight
Наука, що стоїть за активацією Nrf2
Після того, як у 2 році була створена початкова дієтична добавка, що активує Nrf2004, була відома мінімальна інформація про функцію шляху Nrf2. У літературі про Nrf200, також відомому як ядерний фактор 2 або NFE2L2, існувало приблизно 2 газет, і дослідники тільки починали відкривати антиоксидантну реакцію Nrf2 у ссавців. Однак станом на 2017 рік було надруковано понад 9,300 академічних досліджень щодо цього «головного регулятора».
Насправді, Nrf2 регулює багато антиоксидантних ферментів, які не пов’язані з генами, натомість вони пропонують захист від різноманітних обставин, пов’язаних зі стресом, з якими стикаються клітини, органи і, зрештою, організми за здорових і патологічних умов. На основі цієї нової кількості інформації з опублікованих академічних досліджень тепер дослідники можуть розвиватися краще БАД Nrf2.
Станом на 2007 рік дослідницькі дослідження продемонстрували складну функцію шляху Nrf2. Було виявлено, що активатори Nrf2 імітують фактори різних структур в організмі людини. За допомогою цих шляхів активатори Nrf2 були обладнані для того, щоб відчувати зміну умов по всій клітині, щоб підтримувати баланс і реагувати на зростаючі потреби генів.
Навіщо використовувати добавки, що активують Nrf2?
Оскільки здатність організму до активації Nrf2 з віком зменшується, можуть початися зміни. Дослідження продемонстрували, що фокус Nrf2 в клітинах зменшується з віком, демонструючи збільшення маркерів окисного стресу. Через ці зміни можуть розвиватися різноманітні вікові захворювання, такі як атеросклероз і серцево-судинні захворювання, артрит, рак, ожиріння, цукровий діабет 2 типу, гіпертонія, катаракта, хвороба Альцгеймера, а також хвороба Паркінсона. При цих проблемах зі здоров’ям виявлено окислювальний стрес.
Стимулюючи здатність клітини збільшувати вироблення активаторів Nrf2, БАД Nrf2 може допомогти відновити здатність людського організму протидіяти наслідкам окисного стресу. Поліненасичені жирні кислоти, або ПНЖК, є одними з молекул, які найбільш легко окислюються, і вони особливо вразливі до пошкодження від вільних радикалів. Продукція тіобарбітурової кислоти, або TBARS, може збільшуватися з віком, що вказує на підвищений окислювальний стрес разом із зниженням Nrf2-регульованих шляхів.
З біологічної точки зору індукція генів є дуже повільним механізмом, який, як правило, вимагає годин для передачі по шляху. В результаті �багато ферментів мають власні перемикачі включення/вимкнення, які можуть запускатися за лічені хвилини різними регуляторними ферментами. Дослідники розробили запатентовані композиції активаторів Nrf2, які використовують цю базу знань про активацію. Активація Nrf2 складається не тільки з того, що фактор транскрипції Nrf2 вивільняється з інгібітора і мігрує до клітинного ядра, але також зв’язується зі специфічними послідовностями ДНК, щоб стимулювати експресію цитопротекторного гена, регулюючи швидкість виведення Nrf2 з ядра.
Розуміння процедури елімінації та активації Nrf2 в організмі людини дозволило дослідникам створювати комбінації різних активаторів Nrf2, щоб здійснити відображення генів за допомогою його модуляції. Поєднання бази знань разом з широким спектром інших досліджень допомогло створити активатори Nrf2 для використання в якості харчових добавок. Обсяг нашої інформації обмежено хіропрактикою та проблемами здоров’я хребта. Щоб обговорити тему, зверніться до доктора Хіменеса або зв’яжіться з нами за адресою�915-850-0900.
Куратор доктор Алекс Хіменес
Додаткова тема для обговорення: полегшення болю в коліні без операції
Біль у коліні є добре відомим симптомом, який може виникнути через різноманітні травми та/або стани коліна, у тому числі �спортивні травми. Коліно є одним із найскладніших суглобів в організмі людини, оскільки складається з перетину чотирьох кісток, чотирьох зв’язок, різних сухожиль, двох менісків і хрящів. За даними Американської академії сімейних лікарів, найпоширенішими причинами болю в коліні є підвивих колінної чашечки, тендиніт надколінка або коліно стрибуна та хвороба Осгуда-Шлаттера. Хоча біль у колінах найчастіше виникає у людей старше 60 років, біль у колінах також може виникати у дітей та підлітків. Біль у коліні можна лікувати вдома за методами RICE, однак серйозні травми коліна можуть вимагати негайної медичної допомоги, включаючи хіропрактику.
Антиоксиданти науково називають сполуками, які обмежують процес окислення в організмі людини, і якщо їх не контролювати, вони можуть створювати вільні радикали, які можуть розвинути численні ланцюгові реакції, які можуть викликати пошкодження клітин. На щастя, людський організм може створити такі вбудовані імунні механізми, однак, коли активні форми кисню, або АФК, не можуть бути нейтралізовані, уявіть собі крихітне полум’я, яке виходить з-під контролю при наповненні киснем, шкода обов’язково станеться. .
Якщо продовжувати розширювати метафору полум’я, кінцевим продуктом відсутності здатності нейтралізувати вплив АФК, або активних форм кисню, є пошкодження, а також запалення, іншими словами, тіло людини буквально горить. Фантастичне те, що є антиоксиданти, які можуть надзвичайно допомогти в боротьбі з цією проблемою зі здоров’ям, і цим антиоксидантом є глутатіон. Незважаючи на те, що антиоксидантний ефект глутатіону був знайдений у 1889 році, він став однією з найцікавіших тем у сучасних дослідженнях.
Майстер антиоксидантів: глутатіон
Потужна речовина — це трипептид, який утворюється з цистеїну, глутамінової кислоти та гліцину. Завдяки своїй здатності захищати організм людини від утворення вільних радикалів, глутатіон може в кінцевому підсумку сприяти зміцненню здорової імунної системи. На основі Наукові звіти за 2015 рік, було встановлено, що здатність глутатіону функціонувати синергетично з пероксиредином і каталазою допомагає захищати клітини від перекису водню. Ця синергічна формула діє проти активних форм кисню, або АФК. Глутатіон, пероксидредин і каталаза є важливими елементами для підвищення клітинного гомеостазу, який є важливим процесом для здорових клітин, тканин і органів в цілому.
Крім того, глутатіон покращує загальну структуру та функції імунної системи, використовуючи його важливий вплив на функції лімфоцитів. Відповідно з кафедра імунохімії, належне доповнення рівня глутатіону в організмі людини може значно посилити імунні реакції. Як приклад, два рандомізованих плацебо-контрольованих дослідження продемонстрували, що терапевтичне лікування пацієнтів з ослабленим імунітетом N-ацетилцистеїном, або NAC, в обох випадках призвело до значного зростання більшості імунологічних процесів, що включало повне омолодження. активність природних клітин-кілерів. N-ацетил-цистеїн, або NAC, використовує сірку з глутатіону і поєднує її з отруйними молекулами, які потім стають водорозчинними і викидаються в організм людини.
Глутатіон також має здатність відновлювати ліпоєву кислоту, а також переробляти вітаміни С і Е, які необхідні для ініціювання певних системних процесів, посилаючи електрони для нейтралізації вільних радикалів. На основі дослідження з PLoS ONE, глутатіон вражав пацієнтів з метильним цукровим діабетом, або СД2, і мікобактерією туберкульозу. Зазвичай люди зі слабкою імунною системою мають тенденцію до більшого впливу на M. tb, або мікобактерії туберкульозу, хвороби чи інфекції. Крім того, люди з цукровим діабетом 2 типу або СД2 в два-три рази більш схильні до туберкульозу, ніж люди без ЦД 2 типу. Дослідження також показало, що підвищення рівня глутатіону в макрофагах, виділених у пацієнтів із ЦД 2-го типу, призвело до покращення контролю захворювання або інфекції M.Tb. Ці результати демонструють, що нижчі рівні глутатіону у пацієнтів із ЦД2 сприяють підвищенню ймовірності захворювання або інфекції M. tb. Більше того, в залежності від Дієтро Гецці в медичній школі Брайтона та Сассекса, окислювальний стрес може в кінцевому підсумку спричинити погану структуру та роботу імунної системи.
На щастя, глутатіон відіграє важливу роль у зміцненні та контролі імунітету. Наприклад, глутатіон має важливе значення для вроджених і адаптаційних процесів в імунній системі, включаючи проліферацію Т-лімфоцитів, фагоцитарну активність поліморфноядерних нейтрофілів і функції дендритних клітин, які можуть бути фундаментальними, оскільки вони складаються з антигенпрезентуючих клітин. . Клітинний імунітет включає білкові антигени, які спочатку починають дегенерувати в ендоцитарних везикулах макрофагів і дендритних клітин, тому менші пептиди демонструються на поверхні, щоб активувати проліферацію антиген-специфічних Т-клітин. Крім того, глутатіон допомагає у створенні цитокінів, і необхідно підтримувати вироблення інтерферону-гамма дендритними клітинами, що важливо для захисту від внутрішньоклітинних патогенів, включаючи мікобактерії.
N-ацетил-цистеїн, або NAC, науково званий попередником глутатіону, також є дуже потужним клітинним антиоксидантом, який використовується як антиоксидант-поглинач вільних радикалів. Загальновідомий за свою роль у запобігання токсичності ацетамінофенуБуло продемонстровано, що NAC, або N-ацетил-цистеїн, володіє кількома перевагами для здоров’я та самопочуття. Відповідно до Стільниковий журналNAC допомагає підтримувати здорову запальну відповідь і може позитивно вплинути на доношені та передчасні пологи. Дослідження прийшло до висновку, що у жінок з попередніми передчасними пологами та бактеріальним вагінозом 0.6 грама NAC на день, які приймали перорально разом із прогестероном після 16 тижня вагітності, захищали від рецидиву передчасних пологів та покращували неонатальний результат. На завершення також було виявлено позитивний вплив NAC на нарощування м’язової маси. Після трьох хвилин постійних скорочень відбулося збільшення продуктивності на 15 відсотків, що демонструє, як NAC відіграє основну роль у покращенні нарощування м’язової маси та зниженні загальної втоми під час пологів.
Дослідники також виявили, що NAC, або N-ацетил-цистеїн, може бути корисним для тих, у кого є синдром полікістозних яєчників або СПКЯ. СПКЯ, або синдром полікістозних яєчників, є поширеним захворюванням, пов’язаним з ендокринними залозами, яке вражає приблизно від 5 до 10 відсотків жінок репродуктивного віку. У таких пацієнтів існує більший ризик виникнення метаболічного синдрому, коли використання NAC допомогло відновити здоровий рівень інсуліну та чутливість.
Інсайт доктора Алекса Хіменеса
Глутатіон називають «майстером антиоксидантів» через його фундаментальну роль у досягненні та підтримці загального здоров’я та гарного самопочуття. Хоча людський організм здатний виробляти власний глутатіон, погане харчування, забруднення, токсини, надмірне вживання наркотиків та/або ліків, стрес, травми, старіння, хвороби та радіація можуть знизити природний рівень глутатіону. Це, у свою чергу, може зробити людей більш сприйнятливими до пошкодження клітин від окисного стресу, вільних радикалів, інфекцій та раку. Тому добавки глутатіону можуть мати величезну користь для людського організму. Разом з альтернативними методами лікування, такими як хіропрактика, рівень глутатіону можна знову регулювати, щоб покращити самопочуття.
Крім того, медичні працівники запропонували використовувати добавки глутатіону разом з іншими альтернативними методами лікування, такими як хіропрактіка, для подальшого покращення загального здоров’я та самопочуття. Антиоксиданти важливі для підтримки максимального самопочуття, а також для пригнічення ланцюгової реакції вільних радикалів, які завдають шкоди або пошкодження клітин. Потужні антиоксиданти, такі як глутатіон, як згадувалося вище, в кінцевому підсумку допомагають регулювати розвиток цих вільних радикалів і забезпечують більш здорову відповідь імунної системи. Дослідження показали, що хіропрактіка може також відігравати важливу роль у цьому процесі, природним чином підвищуючи активність антиоксидантів в організмі людини. Хіропрактика – це безпечний та ефективний підхід до лікування, який використовує коригування хребта та ручні маніпуляції для виправлення викривлень хребта або підвивиху, щоб дозволити людському тілу природним чином вилікуватися без використання ліків/лікарських засобів та/або хірургічних втручань.
Нарешті, антиоксиданти демонструють свої біологічні властивості через велику кількість користі для здоров’я, яка може бути необхідна зараз, як ніколи раніше, з кожним зростаючим натиском стресів, хвороб і забруднення в нашому сучасному світі, які всі сприяють пошкодженню та/або пошкодженню клітин. . Глутатіон та його попередник NAC, або N-ацетил-цистеїн, продовжують демонструвати свій потужний статус у сфері антиоксидантів. Разом з альтернативними методами лікування, такими як хіропрактика, люди можуть скористатися всіма перевагами, які може запропонувати цей потужний антиоксидант. Обсяг нашої інформації обмежений хіропрактикою, а також травмами та станами хребта. Щоб обговорити тему, зверніться до доктора Хіменеса або зв’яжіться з нами за адресою�915-850-0900.
Куратор доктор Алекс Хіменес
Додаткові теми: Біль у спині
Біль у спині є однією з найпоширеніших причин інвалідності та пропущених робочих днів у всьому світі. Насправді, біль у спині вважається другою за поширеністю причиною відвідувань лікаря, переважаючи лише інфекції верхніх дихальних шляхів. Приблизно 80 відсотків населення відчувають біль у спині принаймні один раз протягом життя. Хребет – це складна структура, що складається з кісток, суглобів, зв’язок і м’язів, а також інших м’яких тканин. Через це травми та/або загострення стану, наприклад грижі диски, може зрештою призвести до симптомів болю в спині. Спортивні травми або травми в автомобільній катастрофі часто є найчастішою причиною болю в спині, однак іноді найпростіші рухи можуть мати хворобливі наслідки. На щастя, альтернативні варіанти лікування, такі як хіропрактика, можуть допомогти полегшити біль у спині за допомогою корекції хребта та ручних маніпуляцій, що в кінцевому підсумку покращує полегшення болю.
Науково обґрунтований мануальний терапевт доктор Олександр Хіменес дивиться на це окислювальний стрес, що це таке, як він впливає на організм і антиоксидантний захист, щоб виправити ситуацію.
Есра Бірбен, доктор медичних наук, 1 Уміт Мурат Сахінер, 1 доктор медичних наук Кансін Сакесен, 1 доктор медичних наук Серпіл Ерзурум, 2 та Омер Калайчі, доктор медичних наук, 1
Анотація: Активні форми кисню (АФК) виробляються живими організмами в результаті нормального клітинного метаболізму та факторів навколишнього середовища, таких як забруднювачі повітря або сигаретний дим. АФК є високореактивними молекулами, які можуть пошкоджувати клітинні структури, такі як вуглеводи, нуклеїнові кислоти, ліпіди та білки, і змінювати їх функції. Зміщення балансу між окислювачами та антиоксидантами на користь окислювачів називається «оксидативним стресом». Регуляція відновного та окислювально-відновного стану є критичною для життєздатності, активації, проліферації та функціонування органів. Аеробні організми мають інтегровані антиоксидантні системи, які включають ферментні та неферментні антиоксиданти, які зазвичай ефективно блокують шкідливі ефекти АФК. Однак при патологічних станах антиоксидантні системи можуть бути перевантажені. Окислювальний стрес сприяє виникненню багатьох патологічних станів і захворювань, включаючи рак, неврологічні розлади, атеросклероз, гіпертензію, ішемію/перфузію, цукровий діабет, гострий респіраторний дистрес-синдром, ідіопатичний фіброз легенів, хронічну обструктивну хворобу легень та астму. У цьому огляді ми підсумовуємо клітинні окислювальні та антиоксидантні системи та обговорюємо клітинні ефекти та механізми окисного стресу.
Ключові слова: антиоксидант, окислювач, окислювальний стрес, активні форми кисню, окислювально-відновний
(WAO Journal 2012; 5:9�19)
Активні форми кисню (АФК) виробляються живими організмами в результаті нормального клітинного метаболізму. У низьких або помірних концентраціях вони функціонують у фізіологічних процесах клітини, але при високих концентраціях вони викликають несприятливі модифікації компонентів клітини, таких як ліпіди, білки та ДНК.1�6 Зміщення балансу між окислювачем/антиоксидантом на користь окисників Окислювальний стрес сприяє розвитку багатьох патологічних станів, включаючи рак, неврологічні розлади, 7�10 атеросклероз, гіпертензію, ішемію/перфузію, 11�14 цукровий діабет, гострий респіраторний дистрес-синдром, ідіопатичний легеневий фіброз, хронічну обструктивну хворобу легень. ,15 і астма.16�21 Аеробні організми мають інтегровані антиоксидантні системи,� які включають ферментні та неферментні антиоксиданти, які зазвичай ефективно блокують шкідливі ефекти АФК. Однак при патологічних станах антиоксидантні системи можуть бути перевантажені. У цьому огляді ми підсумовуємо клітинні окислювальні та антиоксидантні системи та регуляцію відновного та окислювально-відновного (окислювально-відновного) стану у здоров’я та хворобливих станах.
ОКСИДАНТИ
Ендогенні джерела АФК
АФК виробляються з молекулярного кисню в результаті нормального клітинного метаболізму. АФК можна розділити на 2 групи: вільні радикали і нерадикали. Молекули, що містять один або кілька неспарених електронів і таким чином забезпечують реакційну здатність молекулі, називаються вільними радикалами. Коли 2 вільні радикали поділяють свої неспарені електрони, утворюються нерадикальні форми. 3 основних АФК, які мають фізіологічне значення, це супероксид-аніон (O22.), гідроксильний радикал (OH) і перекис водню (H2O2). ROS узагальнено в таблиці 1.
Супероксид-аніон утворюється при додаванні 1 електрона до молекулярного кисню.22 Цей процес опосередковується нікотин-аденін-динуклеотидфосфат [NAD(P)H]-оксидазою або ксантиноксидазою або мітохондріальною системою транспорту електронів. Основним місцем утворення супероксид-аніона є мітохондрії, механізм клітини для виробництва аденозинтрифосфату. Зазвичай електрони передаються через мітохондріальний ланцюг транспорту електронів для відновлення кисню до води, але приблизно від 1 до 3% всіх електронів витікає з системи і виробляє супероксид. NAD(P)H оксидаза міститься в поліморфноядерних лейкоцитах, моноцитах і макрофагах. Після фагоцитозу ці клітини виробляють сплеск супероксиду, який призводить до бактерицидної активності. Супероксид перетворюється на перекис водню під дією супероксиддисмутаз (СОД, ЕС 1.15.1.1). Перекис водню легко дифундує через плазматичну мембрану. Перекис водню також виробляється ксантиноксидазою, оксидазою амінокислот і NAD(P)H оксидазою23,24 і в пероксисомах шляхом споживання молекулярного кисню в метаболічних реакціях. У послідовності реакцій, які називаються реакціями Габера-Вейса і Фентона, H2O2 може розпадатися до OH2 у присутності пропускаючих металів, таких як Fe21 або Cu21.25.
Fe31 +�.O2�?Fe2 +�O2 Габер Вайс
Fe2 +�H2O2�?Fe3 +�OH�+ .OH Реакція Фентона
Сам O 2 також може реагувати з H2 O2 і утворювати OH.26,27 Гідроксильний радикал є найбільш реакційноздатним з АФК і може пошкоджувати білки, ліпіди, вуглеводи та ДНК. Він також може почати перекисне окислення ліпідів, забираючи електрон з поліненасичених жирних кислот.
Гранульоцитарні ферменти додатково посилюють реакційну здатність H2O2 за допомогою пероксидази еозинофілів і мієлопероксидази (MPO). В активованих нейтрофілах H2O2 споживається МПО. У присутності хлорид-іона H2O2 перетворюється на хлорноватисту кислоту (HOCl). HOCl має високу окислювальну дію і відіграє важливу роль у знищенні патогенів у дихальних шляхах.28 Однак HOCl також може реагувати з ДНК та індукувати взаємодії ДНК з білками та виробляти продукти окислення піримідину та додавати хлорид до основ ДНК.29,30 Пероксидаза еозинофілів. і MPO також сприяють окисному стресу шляхом модифікації білків галогенуванням, нітруванням і поперечними зв'язками білків через тирозилові радикали.31�33
Іншими вільними радикалами, що отримують кисень, є пероксильні радикали (ROO$). Найпростішою формою цих радикалів є гідропероксильний радикал (HOO$), який бере участь у перекисному окисленні жирних кислот. Вільні радикали можуть викликати ланцюгові реакції перекисного окислення ліпідів шляхом відриву атома водню від вуглецю метилену бічного ланцюга. Потім ліпідний радикал реагує з киснем, утворюючи пероксильний радикал. Пероксильний радикал ініціює ланцюгову реакцію і перетворює поліненасичені жирні кислоти в гідропероксиди ліпідів. Гідропероксиди ліпідів дуже нестабільні і легко розкладаються на вторинні продукти, такі як альдегіди (наприклад, 4-гідрокси-2,3-ноненаль) і малоновий діальдегід (MDA). Ізопростани є іншою групою продуктів перекисного окислення ліпідів, які утворюються в результаті перекисного окислення арахідонової кислоти, а також виявлено, що вони підвищені в плазмі та в конденсаті дихання астматиків.34,35 Перекисне окислення ліпідів порушує цілісність клітинних мембран і призводить до перебудови структура мембрани.
Перекис водню, супероксидний радикал, окислений глутатіон (GSSG), MDA, ізопростани, карбоніли та нітротирозин можна легко виміряти із зразків плазми, крові або бронхоальвеолярного лаважу як біомаркери окислення за допомогою стандартизованих аналізів.
Екзогенне джерело окисників
Сигаретний дим
Сигаретний дим містить багато окислювачів і вільних радикалів і органічних сполук, таких як супероксид і оксид азоту.36 Крім того, вдихання сигаретного диму в легені також активує деякі ендогенні механізми, такі як накопичення нейтрофілів і макрофагів, які ще більше посилюють окислювальне ураження. .
Вплив озону
Вплив озону може викликати перекисне окислення ліпідів і спричинити приплив нейтрофілів в епітелій дихальних шляхів. Короткочасний вплив озону також викликає вивільнення медіаторів запалення, таких як MPO, еозинофільні катіонні білки, а також лактатдегідрогеназа та альбумін.37 Навіть у здорових людей вплив озону спричиняє зниження легеневих функцій.38 Cho et al39 показали, що тверді частинки (суміш твердих частинок і рідких крапель, зважених у повітрі) каталізують відновлення кисню.
Гіпероксия
Гіпероксия відноситься до станів з більш високим рівнем кисню, ніж нормальний парціальний тиск кисню в легенях або інших тканинах тіла. Це призводить до більшого виробництва активних форм кисню та азоту.40,41
Іонізуюче випромінювання
Іонізуюче випромінювання в присутності О2 перетворює гідроксильний радикал, супероксид і органічні радикали в перекис водню і органічні гідропероксиди. Ці види гідропероксиду реагують з окислювально-відновними активними іонами металів, такими як Fe і Cu, за допомогою реакцій Фентона і, таким чином, викликають окислювальний стрес.42,43 Narayanan et al44 показали, що фібробласти, які піддавалися впливу альфа-частинок, мали значне збільшення внутрішньоклітинного O2 2 і H2O2 вироблення через зв'язану з плазматичною мембраною NADPH оксидазу.44 Молекули передачі сигналу, такі як позаклітинна кіназа 1 і 2, що регулюється сигналом (ERK1/2), N-кіназа c-Jun (JNK) і p38, а також фактори транскрипції, такі як Білок-активатор-1 (AP-1), ядерний фактор-kB (NF-kB) і p53 активуються, що призводить до експресії генів, пов’язаних з радіаційною реакцією.45�50 Ультрафіолетові фотони A (UVA) запускають окислювальні реакції шляхом збудження ендогенних фотосенсибілізаторів, таких як порфірини, НАДФН оксидаза і рибофлавіни. 8-Oxo-7,8-дігідрогуанін (8-oxoGua) є основним продуктом окислення ДНК, опосередкованим УФА, утвореним окисленням радикала OH, 1-електронних окисників і синглетного кисню, який в основному реагує з гуаніном.51 Утворення гуаніну Було показано, що катіон-радикал в ізольованій ДНК ефективно виникає завдяки прямому впливу іонізуючого випромінювання.52,53 Після впливу іонізуючого випромінювання внутрішньоклітинний рівень глутатіону (GSH) знижується на короткий термін, але потім знову зростає.54
Іони важких металів
Іони важких металів, такі як залізо, мідь, кадмій, ртуть, нікель, свинець і миш'як, можуть викликати утворення реактивних радикалів і викликати пошкодження клітин через виснаження активності ферментів через перекисне окислення ліпідів і реакцію з ядерними білками та ДНК.55
Одним з найважливіших механізмів утворення вільних радикалів, опосередкованих металами, є реакція типу Фентона. Супероксид-іон і перекис водню можуть взаємодіяти з перехідними металами, такими як залізо і мідь, через каталізовану металом реакцію Габера-Вейса/Фентона, утворюючи радикали OH.
Крім механізмів типу Фентона і Габера-Вейса, певні іони металів можуть реагувати безпосередньо з клітинними молекулами, утворюючи вільні радикали, такі як тіолові радикали, або індукувати клітинні сигнальні шляхи. Ці радикали також можуть реагувати з іншими молекулами тіолу з утворенням O22.. O22. перетворюється на H2O2, що викликає додаткове утворення радикалів кисню. Деякі метали, такі як арсеніт, індукують утворення АФК опосередковано шляхом активації систем, що продукують радикали в клітинах.56
Миш'як є високотоксичним елементом, який продукує різноманітні АФК, включаючи супероксид (O2 2), синглетний кисень (1O2), пероксильний радикал (ROO), оксид азоту (NO), перекис водню (H2O2) та пероксильні радикали диметиларсину [( CH3)2AsOO].57�59 Сполуки миш'яку (III) можуть інгібувати антиоксидантні ферменти, особливо GSH-залежні ферменти, такі як глутатіон-S-трансферази (GSTs), глутатіонпероксидаза (GSH-Px) і GSH-редуктаза, через зв'язування - до їх сульфгідрильних (�SH) груп.60,61
Свинець збільшує перекисне окислення ліпідів.62 Повідомлялося про значне зниження активності тканинної SOD і GPx мозку після впливу свинцю.63,64 Заміна цинку, який служить кофактором для багатьох ферментів, свинцем призводить до інактивації таких ферментів. Вплив свинцю може спричинити пригнічення GST шляхом впливу на тканинні тіоли.
АФК, що утворюється в реакціях, що каталізуються металами, можуть модифікувати основи ДНК. Три заміни основ, G / C, G / T і C / T, можуть виникнути в результаті окисного пошкодження іонами металів, такими як Fe21, Cu21 і Ni21. Reid et al65 показали, що G/C переважно вироблявся Fe21, тоді як C/T заміщення відбувалося Cu21 та Ni21.
АНТИОКСИДАНТИ
Організм людини забезпечений різноманітними антиоксидантами, які служать для противаги дії окислювачів. Для всіх практичних цілей їх можна розділити на 2 категорії: ферментативні (табл. 2) і неферментні (табл. 3).
Ферментні антиоксиданти
Основними ферментними антиоксидантами легенів є СОД (EC 1.15.1.11), каталаза (EC 1.11.1.6) і GSH-Px (EC 1.11.1.9). На додаток до цих основних ферментів, інші антиоксиданти, включаючи гемоксигеназу-1 (EC 1.14.99.3), і окислювально-відновні білки, такі як тіоредоксини (TRXs, EC 1.8.4.10), пероксиредоксини (PRXs, EC 1.11.1.15) і глутаредоксини Також було виявлено, що вони відіграють вирішальну роль у захисті легенів від антиоксидантів.
Оскільки супероксид є основною АФК, що виробляється з різних джерел, його дисмутація за допомогою СОД має першочергове значення для кожної клітини. Усі 3 форми СОД, тобто CuZn-SOD, Mn-SOD і EC-SOD, широко експресуються в легенях людини. Mn-SOD локалізується в матриксі мітохондрій. EC-SOD в основному локалізується в позаклітинному матриксі, особливо в областях, що містять велику кількість колагенових волокон типу I, а також навколо легеневих і системних судин. Він також був виявлений в бронхіальному епітелії, альвеолярному епітелії та альвеолярних макрофагах.66,67 Загалом вважають, що CuZn-SOD і Mn-SOD діють як масові поглиначі супероксидних радикалів. Відносно високий рівень EC-SOD в легенях з його специфічним зв’язуванням з компонентами позаклітинного матриксу може бути основним компонентом захисту легеневого матриксу.68
H2O2, що утворюється під дією SOD або дії оксидаз, таких як ксантиноксидаза, відновлюється до води за допомогою каталази та GSH-Px. Каталаза існує як тетрамер, що складається з 4 ідентичних мономерів, кожен з яких містить групу гема в активному центрі. Деградація H2O2 здійснюється шляхом перетворення між 2 конформаціями каталази-ферикаталази (залізо координується з водою) і сполуки I (залізо в комплексі з атомом кисню). Каталаза також зв’язує NADPH як відновний еквівалент для запобігання окисної інактивації ферменту (утворення сполуки II) H2O2, коли він відновлюється до води.69
Ферменти окислювально-відновного циклу, що відповідають за відновлення H2O2 і гідропероксидів ліпідів (утворених в результаті перекисного окислення ліпідів мембрани), включають GSH-Pxs.70 GSH-Pxs – це сімейство тетрамерних ферментів, які містять унікальну амінокислоту селеноцистеїн в активних центрів і використовують низькомолекулярні тіоли, такі як GSH, для відновлення H2O2 і пероксидів ліпідів до відповідних спиртів. Було описано чотири GSH-Px, які кодуються різними генами: GSH-Px-1 (клітинний GSH-Px) поширений повсюдно і зменшує кількість H2O2 і пероксидів жирних кислот, але не етерифіковані пероксиліпіди.71 Етерифіковані ліпіди відновлюються мембранно-зв’язаним GSH. -Px-4 (фосфоліпідний гідропероксид GSH-Px), який може використовувати кілька різних низькомолекулярних тіолів як відновлювальні еквіваленти. GSH-Px-2 (шлунково-кишковий GSH-Px) локалізується в епітеліальних клітинах шлунково-кишкового тракту, де він служить для зменшення вмісту пероксидів у їжі.72 GSH-Px-3 (позаклітинний GSH-Px) є єдиним членом сімейства GSH-Px, який знаходиться в позаклітинний компартмент і вважається одним з найважливіших позаклітинних антиоксидантних ферментів у ссавців. З них позаклітинний GSH-Px найбільш широко досліджується в легенях людини.73
Крім того, видалення H2O2 тісно пов’язане з кількома тіоловмісними ферментами, а саме, TRXs (TRX1 і TRX2), тіоредоксинредуктазами (EC 1.8.1.9) (TRRs), PRXs (які є тіоредоксинпероксидазами) та глутаредоксинами.
Два TRX і TRR були охарактеризовані в клітинах людини, які існують як в цитозолі, так і в мітохондріях. У легенях TRX і TRR експресуються в бронхіальному та альвеолярному епітелії та макрофагах. У клітинах людини виявлено шість різних PRX, які відрізняються своєю ультраструктурною компартментальністю. Експериментальні дослідження виявили важливість PRX VI для захисту альвеолярного епітелію. Легені людини експресують всі PRX в бронхіальному епітелії, альвеолярному епітелії та макрофагах.75 Нещодавно було встановлено, що PRX V функціонує як пероксинітрит-редуктаза,76 що означає, що він може функціонувати як потенційна захисна сполука при розвитку опосередкованого АФК ураження легень. .77
Загальним для цих антиоксидантів є потреба в НАДФН як відновного еквівалента. NADPH підтримує каталазу в активній формі і використовується як кофактор TRX і GSH редуктазою (EC 1.6.4.2), яка перетворює GSSG в GSH, ко-субстрат для GSH-Pxs. Внутрішньоклітинний НАДФН, у свою чергу, утворюється внаслідок відновлення NADP1 глюкозо-6-фосфатдегідрогеназою, першим і лімітуючим швидкість ферменту пентозофосфатного шляху, під час перетворення глюкозо-6-фосфату в 6-фосфоглюконолактон. Утворюючи NADPH, глюкозо-6-фосфатдегідрогеназа є критичною детермінантою буферної здатності цитозольного GSH (GSH/GSSG) і, отже, може вважатися важливим, регуляторним антиоксидантним ферментом.78,79
GST (EC 2.5.1.18), інше сімейство антиоксидантних ферментів, інактивує вторинні метаболіти, такі як ненасичені альдегіди, епоксиди та гідропероксиди. Було описано три основні сімейства GST: цитозольний GST, мітохондріальний GST, 80,81 і асоційований з мембраною мікросомальний GST, який відіграє роль в метаболізмі ейкозаноїдів і GSH.82 У ссавців ідентифіковано сім класів цитозольних GST, позначених Alpha, Mu, Pi, Sigma, Theta, Omega і Zeta.83�86 Під час ненапружених умов GST класу Mu і Pi взаємодіють з кіназами Ask1 і JNK, відповідно, і інгібують ці кінази.87�89 Було показано, що GSTP1 дисоціює з JNK у відповідь на окислювальний стрес.89 GSTP1 також фізично взаємодіє з PRX VI і призводить до відновлення активності ферменту PRX через глутатіонілування окисленого білка.90
Неферментні антиоксиданти
До неферментативних антиоксидантів належать низькомолекулярні сполуки, такі як вітаміни (вітаміни C і E), b-каротин, сечова кислота і GSH, трипептид (Lg-глутаміл-L-цистеїніл-L-гліцин), який містить тіол ( сульфгідрильна) група.
Вітамін С (аскорбінова кислота)
Водорозчинний вітамін С (аскорбінова кислота) забезпечує внутрішньоклітинну та позаклітинну водно-фазну антиоксидантну здатність, головним чином, поглинаючи вільні радикали кисню. Він перетворює вільні радикали вітаміну Е назад у вітамін Е. Було показано, що рівень його в плазмі знижується з віком.91,92
Вітамін Е (а-токоферол)
Ліпідорозчинний вітамін Е зосереджений у гідрофобній внутрішній частині клітинної мембрани і є основним захистом від пошкодження мембрани, викликаного окисниками. Вітамін Е віддає електрон пероксильному радикалу, який утворюється під час перекисного окислення ліпідів. a-токоферол є найактивнішою формою вітаміну Е та основним мембранно-зв'язаним антиоксидантом у клітині. Вітамін Е запускає апоптоз ракових клітин і пригнічує утворення вільних радикалів.93
Глутатіон
GSH у великій кількості міститься у всіх клітинних компартментах і є основним розчинним антиоксидантом. Співвідношення GSH/GSSG є основним фактором, що визначає окислювальний стрес. GSH проявляє свою антиоксидантну дію кількома способами.94 Він детоксикує перекис водню та перекиси ліпідів за допомогою дії GSH-Px. GSH віддає свій електрон H2O2, щоб відновити його в H2O та O2. GSSG знову відновлюється в GSH за допомогою GSH-редуктази, яка використовує NAD(P)H як донор електронів. GSH-Pxs також важливі для захисту клітинної мембрани від перекисного окислення ліпідів. Відновлений глутатіон віддає протони мембранним ліпідам і захищає їх від атак окислювача.95
GSH є кофактором для кількох детоксикаційних ферментів, таких як GSH-Px та трансферази. Він бере участь у перетворенні вітамінів С і Е назад у активні форми. GSH захищає клітини від апоптозу, взаємодіючи з проапоптотичними та антиапоптотичними сигнальними шляхами.94 Він також регулює та активує декілька факторів транскрипції, таких як AP-1, NF-kB та Sp-1.
Каротиноїди (b-каротин)
Каротиноїди - це пігменти, що містяться в рослинах. В першу чергу було виявлено, що b-каротин реагує з пероксильними (ROO), гідроксильними (OH) і супероксидними (O22.) радикалами.96 Каротиноїди проявляють антиоксидантну дію при низькому парціальному тиску кисню, але можуть мати прооксидантний ефект при більш високому кисні. концентрації.97 Як каротиноїди, так і ретиноєва кислота (РА) здатні регулювати фактори транскрипції.98 b-каротин пригнічує активацію NF-kB, індуковану окислювачем, і вироблення інтерлейкіну (IL)-6 та фактора некрозу пухлини-a. Каротиноїди також впливають на апоптоз клітин. Антипроліферативний ефект РА було показано в кількох дослідженнях. Цей ефект РА опосередковується в основному рецепторами ретиноєвої кислоти і варіюється в залежності від типу клітин. Було показано, що в клітинах карциноми молочної залози рецептор ретиноєвої кислоти викликає пригнічення росту, індукуючи зупинку клітинного циклу, апоптоз або обидва.99,100
ДІЯ ОКИСНЮВАННЯ СТРЕСУ: ГЕНЕТИЧНІ, ФІЗІОЛОГІЧНІ ТА БІОХІМІЧНІ МЕХАНІЗМИ
Окислювальний стрес виникає, коли баланс між антиоксидантами та АФК порушується через виснаження антиоксидантів або накопичення АФК. Коли виникає окислювальний стрес, клітини намагаються протидіяти окислювальному ефекту та відновити окислювально-відновний баланс шляхом активації або приглушення генів, що кодують захисні ферменти, транскрипційні фактори та структурні білки.101,102 Співвідношення між окисленим і відновленим глутатіоном (2GSH/GSSG) становить один з важливих детермінант окисного стресу в організмі. Більш висока продукція АФК в організмі може змінити структуру ДНК, призвести до модифікації білків і ліпідів, активації кількох спричинених стресом факторів транскрипції та продукції прозапальних і протизапальних цитокінів.
Вплив окисного стресу на ДНК
АФК може призвести до модифікацій ДНК кількома способами, що включає деградацію основ, розриви одно- або дволанцюгової ДНК, пуринові, піримідинові або пов’язані з цукром модифікації, мутації, делеції або транслокації та перехресне зшивання з білками. Більшість із цих модифікацій ДНК (рис. 1) мають велике значення для канцерогенезу, старіння та нейродегенеративних, серцево-судинних та аутоімунних захворювань. Тютюновий дим, окислювально-відновні метали та невідновні метали, такі як залізо, кадмій, хром і миш’як, також беруть участь у канцерогенезі та старінні шляхом утворення вільних радикалів або зв’язування з тіоловими групами. Утворення 8-OH-G є найбільш відомим пошкодженням ДНК, що виникає внаслідок окисного стресу, і є потенційним біомаркером для канцерогенезу.
Промоторні ділянки генів містять консенсусні послідовності для факторів транскрипції. Ці сайти зв’язування фактора транскрипції містять збагачені GC послідовності, які чутливі до атак окислювача. Утворення ДНК 8-OH-G в сайтах зв'язування факторів транскрипції може змінити зв'язування факторів транскрипції і таким чином змінити експресію споріднених генів, як було показано для цільових послідовностей AP-1 і Sp-1 Крім 103-OH-G, Також було показано, що 8-цикло-8,59-дезоксиаденозин (цикло-dA) інгібує транскрипцію з репортерного гена в клітинній системі, якщо він знаходиться в коробці TATA.29 TATA-зв’язуючий білок ініціює транскрипцію, змінюючи згин ДНК. . Зв’язування ТАТА-зв’язуючого білка може бути порушено присутністю цикло-dA.
Окислювальний стрес викликає нестабільність мікросателітних (коротких тандемних повторів) областей. Окисно-відновні активні іони металів, гідроксильні радикали збільшують нестабільність мікросателітів.105 Незважаючи на те, що клітини легко переносять розриви одноланцюгової ДНК, спричинені окислювальним пошкодженням, дволанцюгові розриви ДНК, викликані іонізуючим випромінюванням, можуть бути значною загрозою для виживання клітини.106
Метилування на острівцях CpG в ДНК є важливим епігенетичним механізмом, який може призвести до приглушення генів. Окислення 5-MeCyt до 5-гідроксиметилурацилу (5-OHMeUra) може відбуватися за допомогою реакцій дезамінування/окислення тиміну або проміжних продуктів 5-гідроксиметилцитозину.107 На додаток до модулюючої експресії генів, метилювання ДНК також впливає на організацію хроматину. Аберантні моделі метилювання ДНК, індуковані окислювальними атаками, також впливають на активність відновлення ДНК.
Вплив окисного стресу на ліпіди
АФК може індукувати перекисне окислення ліпідів і порушувати розташування мембранного ліпідного двошарового шару, що може інактивувати пов’язані з мембраною рецептори та ферменти та підвищити проникність тканин.109 Продукти перекисного окислення ліпідів, такі як MDA та ненасичені альдегіди, здатні інактивувати багато клітинних білків, утворюючи перехресні протеїни. -зв'язки.110�112 4-Гідрокси-2-ноненал викликає виснаження внутрішньоклітинного GSH та індукує продукцію пероксиду,113,114 активує рецептор епідермального фактора росту,115 і індукує вироблення фібронектину.116 Продукти перекисного окислення ліпідів, такі як ізобарбитова кислота, реактивна речовина , були використані як непрямі біомаркери окислювального стресу, а підвищені рівні були показані в конденсаті видихуваного повітря або бронхоальвеолярному лаважу або легенях пацієнтів з хронічною обструктивною хворобою легень або курців.117�119
Вплив окисного стресу на білки
АФК може викликати фрагментацію пептидного ланцюга, зміну електричного заряду білків, перехресне зшивання білків та окислення специфічних амінокислот і, отже, призвести до підвищеної сприйнятливості до протеолізу шляхом деградації специфічними протеазами.120 Залишки цистеїну та метіоніну в білках є особливо більш чутливі до окислення.121 Окислення сульфгідрильних груп або метіонінових залишків білків спричиняє конформаційні зміни, розгортання та деградацію білка.8,121–123 Ферменти, у яких метали знаходяться на активних центрах або поблизу них, особливо чутливі до окислення, що каталізується металами. Було показано, що окислювальна модифікація ферментів пригнічує їх діяльність.124,125
У деяких випадках може мати місце специфічне окислення білків. Наприклад, метіонін може бути окислений сульфоксидом метіоніну126 і фенілаланіну до о-тирозину127; сульфгідрильні групи можна окислювати з утворенням дисульфідних зв’язків128, а карбонільні групи можуть бути введені в бічні ланцюги білків. Гамма-промені, окислення, що каталізується металами, HOCl та озон можуть спричинити утворення карбонільних груп.129
Вплив окисного стресу на передачу сигналу
АФК може індукувати експресію кількох генів, які беруть участь у передачі сигналу.1,130 Високе співвідношення для GSH/GSSG важливо для захисту клітини від окисного пошкодження. Порушення цього співвідношення викликає активацію редокс-чутливих факторів транскрипції, таких як NF-kB, AP-1, ядерний фактор активованих Т-клітин і фактор 1, індукований гіпоксією, які беруть участь у запальній відповіді. Активація факторів транскрипції через АФК досягається за допомогою каскадів сигнальної трансдукції, які передають інформацію ззовні всередину клітини. Рецептори тирозинкінази, більшість рецепторів фактора росту, таких як рецептор епідермального фактора росту, рецептор фактора росту ендотелію судин і рецептор фактора росту тромбоцитів, протеїн-тирозинфосфатази та серин/треонін-кінази, є мішенями ROS.131�133 Позаклітинні кінази, що регулюються сигналом, JNK і p38, які є членами сімейства протеїнкіназ, активованих мітогеном, і беруть участь у кількох процесах у клітині, включаючи проліферацію, диференціацію та апоптоз, також можуть регулюватися окисниками.
В умовах окисного стресу залишки цистеїну в ділянці зв’язування ДНК c-Jun, деякі субодиниці AP-1 та інгібуюча kB кіназа піддаються оборотній S-глутатіоляції. Повідомляється, що глутаредоксин і TRX відіграють важливу роль у регуляції окислювально-відновних чутливих сигнальних шляхів, таких як NF-kB і AP-1, протеїнкіназа, активована мітогеном p38, і JNK.134�137
NF-kB може бути активований у відповідь на умови окисного стресу, такі як АФК, вільні радикали та УФ-опромінення.138 Фосфорилювання IkB звільняє NF-kB і дозволяє йому проникати в ядро для активації транскрипції гена.139 Деякі кінази мають повідомлялося про фосфорилування IkB на залишках серину. Ці кінази є мішенями окислювальних сигналів для активації NF-kB.140 Редуценти посилюють зв’язування NF-kB ДНК, тоді як окислювачі інгібують зв’язування ДНК NF-kB. TRX може здійснювати 2 протилежні дії в регуляції NF-kB: у цитоплазмі він блокує деградацію IkB та інгібує активацію NF-kB, але посилює зв’язування NF-kB ДНК у ядрі.141 Активація NF-kB через деградацію, пов’язану з окисленням IkB призводить до активації кількох генів, пов’язаних з антиоксидантним захистом. NF-kB регулює експресію кількох генів, які беруть участь в імунній відповіді, таких як IL-1b, IL-6, фактор некрозу пухлини-a, IL-8 та декілька молекул адгезії.142,143 NF-kB також регулює ангіогенез та проліферацію та диференціювання клітин.
AP-1 також регулюється окислювально-відновним станом. У присутності H2O2 деякі іони металів можуть викликати активацію AP-1. Збільшення співвідношення GSH/GSSG посилює зв’язування AP-1, тоді як GSSG інгібує зв’язування ДНК AP-1.144. Зв’язування ДНК гетеродимеру Fos/Jun збільшується за рахунок зменшення одного консервованого цистеїну в ДНК-зв’язуючому домені кожного з білки,145 у той час як зв’язування ДНК AP-1 може бути інгібовано GSSG у багатьох типах клітин, що свідчить про те, що утворення дисульфідного зв’язку залишками цистеїну інгібує зв’язування з ДНК AP-1 Передача сигналу через окислювальний стрес узагальнена на малюнку 146,147.
ВИСНОВКИ
Окислювальний стрес може виникнути внаслідок надлишкового виробництва АФК метаболічними реакціями, які використовують кисень і зміщують баланс між окислювач/антиоксидант статуси на користь окисників. АФК виробляються в результаті метаболічної діяльності клітин і факторів навколишнього середовища, таких як забруднювачі повітря або сигаретний дим. АФК є високореактивними молекулами через неспарені електрони в їх структурі та реагують з кількома біологічними макромолекулами в клітині, такими як вуглеводи, нуклеїнові кислоти, ліпіди та білки, і змінюють їх функції. АФК також впливає на експресію кількох генів шляхом підвищення регуляції редокс-чутливих факторів транскрипції та ремоделювання хроматину через зміну ацетилювання/деацетилювання гістонів. Регулювання окислювально-відновного стану має вирішальне значення для життєздатності клітин, їх активації, проліферації та функціонування органів.
Посилання
1. Валько М., Роудс К.Дж., Монкол Дж., Ізакович М., Мазур М. Вільні радикали, метали та антиоксиданти при раку, спричиненому окислювальним стресом. Chem Biol Interact. 2006;160:1�40.
2. Halliwell B, Gutterridge JMC. Вільні радикали в біології та медицині. 3-е вид. Нью-Йорк: Oxford University Press; 1999.
3. Марнетт Л.Й. Перекисне окислення ліпідів і пошкодження ДНК малоновим діальдегідом. Mutat Res. 1999;424:83�95.
4. Siems WG, Grune T, Esterbauer H. Утворення 4-гідроксиноненалу під час ішемії та реперфузії тонкої кишки щурів. �Наука про життя 1995;57:785�789.
5. Штадтман Є.Р. Роль окисних видів у старінні. Curr Med Chem. 2004;11:1105�1112.
6. Wang MY, Dhingra K, Hittelman WN, Liehr JG, deAndrade M, Li DH. Передбачувані аддукти ДНК малонового діальдегіду, спричинені перекисним окисленням ліпідів, у тканинах грудей людини. Біомаркери епідеміолу раку Поперед. 1996; 5: 705 710.
7. Дженнер П. Окислювальний стрес при хворобі Паркінсона. Енн Нейрол. 2003;53: S26�S36.
8. Лірас Л., Кернс Нью-Джерсі, Дженнер А., Дженнер П., Холлівелл Б. Оцінка окислювального пошкодження білків, ліпідів і ДНК у мозку пацієнтів з хворобою Альцгеймера. J Neurochem. 1997;68:2061�2069.
9. Сейр Л.М., Сміт М.А., Перрі Г. Хімія та біохімія окисного стресу при нейродегенеративних захворюваннях. Curr Med Chem. 2001;8:721�738.
10. Toshniwal PK, Zarling EJ. Докази підвищення перекисного окислення ліпідів при розсіяному склерозі. Neurochem Res. 1992;17:205�207.
11. Dhalla NS, Temsah RM, Netticadan T. Роль окисного стресу в серцево-судинних захворюваннях. J Гіпертоніки. 2000;18:655�673.
12. Каспарова С., Брезова В., Валько М., Горецький Я., Млинарик В. та ін. Вивчення окисного стресу на моделі хронічної гіпоперфузії мозку у щурів. Neurochem Int. 2005;46:601�611.
13. Kerr S, Brosnan MJ, McIntyre M, Reid JL, Dominiczak AF, Hamilton CA. Продукція супероксид-аніона збільшується в моделі генетичної гіпертензії: роль ендотелію. Гіпертонія. 1999;33:1353�1358.
14. Кукрея Р.С., Гесс М.Л. Вільно-радикальна система кисню: від рівнянь через взаємодію білків мембрани до серцево-судинної травми та захисту. Cardiovasc Res. 1992;26:641�655.
15. Asami S, Manabe H, Miyake J, Tsurudome Y, Hirano T та ін. Куріння сигарет викликає збільшення окисного пошкодження ДНК, 8-гідроксидеоксигуанозину, в центральному відділі легенів людини. Канцерогенез. 1997;18:1763�1766.
16. Андреадіс AA, Hazen SL, Comhair SA, Erzurum SC. Окисні та нітрозативні явища при астмі. Free Radic Biol Med. 2003;35:213�225.
17. Comhair SA, Ricci KS, Arroliga M, Lara AR, Dweik RA та ін. Кореляція системної недостатності супероксиддисмутази з обструкцією повітряного потоку при астмі. Am J Respir Crit Care Med. 2005;172:306�313.
18. Comhair SA, Xu W, Ghosh S, Thunnissen FB, Almasan A та ін. Інактивація супероксиддисмутази в патофізіології ремоделювання та реактивності дихальних шляхів при астмі. Am J Pathol. 2005;166:663�674.
19. Dut R, Dizdar EA, Birben E, Sackesen C, Soyer OU, Besler T, Kalayci O. Окислювальний стрес та його детермінанти в дихальних шляхах дітей з астмою. Алергія. 2008;63:1605�1609.
20. Еркан Г., Бірбен Е., Діздар Е.А., Кескін О., Карааслан С. та ін. Окислювальний стрес та генетичні та епідеміологічні детермінанти окислювального ураження при дитячій астмі. J Allergy Clin Immunol. 2006;118:1097�1104.
21. Фіцпатрик А.М., Тіг Р.Г., Ольгін Ф., Є М., Браун Л.А. Програма дослідження важкої астми. Гомеостаз глутатіону дихальних шляхів змінений у дітей з тяжкою астмою: докази окисного стресу. J Allergy Clin Immunol. 2009;123:146�152.
22. Міллер DM, Buettner GR, Aust SD. Перехідні метали як каталізатори реакцій «автоокислення». Free Radic Biol Med. 1990;8:95�108.
23. Dupuy C, Virion A, Ohayon R, Kaniewski J, D'me D, Pommier J. Механізм утворення перекису водню, що каталізується NADPH оксидазою в плазматичній мембрані щитовидної залози. J Biol Chem. 1991;266:3739�3743.
24. Грейнджер Д.Н. Роль ксантиноксидази та гранулоцитів в ішеміо-перфузійному ураженні. Am J Physiol. 1988;255:H1269�H1275.
25. Фентон HJH. Окислення винної кислоти в присутності заліза. J Chem Soc. 1984;65:899�910.
26. Хабер Ф., Вайс Дж. Дж. Каталітичне розкладання перекису водню солями заліза. Proc R Soc Lond Ser A. 1934; 147:332�351.
27. Ліочов С.І., Фрідович І. Цикл Габер-Вайса 70 років потому: альтернативний погляд. Redox Rep. 2002; 7: 55�57.
28. Клебанов С.Й. Мієлопероксидаза: друг і ворог. J Leukoc Biol. 2005;77:598�625.
29. Уайтман М., Дженнер А., Холлівелл Б. Модифікації основ, викликані хлорноватистой кислотою, в ізольованій ДНК тимуса теляти. Chem Res Toxicol. 1997;10:1240�1246.
30. Кульчарик П.А., Гайнеке Ю.В. Хлорноватиста кислота, що виробляється мієлопероксидазною системою фагоцитів людини, індукує ковалентні поперечні зв’язки між ДНК і білком. Біохімія. 2001;40:3648�3656.
31. Brennan ML, Wu W, Fu X, Shen Z, Song W та ін. Розповідь про дві суперечки: визначення ролі пероксидаз у утворенні нітротирозину in vivo за допомогою мишей з дефіцитом еозинофілів і мієлопероксидази, а також природи реактивних видів азоту, що утворюються пероксидазою. J Biol Chem. 2002;277:17415�17427.
32. Denzler KL, Borchers MT, Crosby JR, Cieslewicz G, Hines EM та ін. Екстенсивна дегрануляція еозинофілів та окислення білків дихальних шляхів, опосередковане пероксидазою, не відбуваються в моделі запалення легенів із проблемою яйцеклітини миші. J Immunol. 2001;167:1672�1682.
33. Ван Дален С.Дж., Вінтерборн К.С., Сентілмохан Р., Кеттл А.Д. Нітрит як субстрат та інгібітор мієлопероксидази. Вплив на нітрування та утворення хлорноватистой кислоти в місцях запалення. J Biol Chem. 2000;275:11638�11644.
34. Вуд Л.Г., Фіцджеральд Д.А., Гібсон П.Г., Купер Д.М., Гарг М.Л. Перекисне окислення ліпідів, визначене ізопростанами плазми, пов’язане з тяжкістю захворювання при легкій астмі. Ліпіди. 2000;35:967-974.
35. Montuschi P, Corradi M, Ciabattoni G, Nightingale J, Haritonov SA, Barnes PJ. Підвищення 8-ізопростану, маркера окисного стресу, у видихуваному конденсаті хворих на астму. Am J Respir Crit Care Med. 1999;160:216�220.
36. Церква Д.Ф., Приор В.А. Вільно-радикальна хімія сигаретного диму та її токсикологічні наслідки. Перспектива здоров'я навколишнього середовища. 1985;64:111�126.
37. Hiltermann JT, Lapperre TS, van Bree L, Steerenberg PA, Brahim JJ та ін. Запалення, спричинене озоном, оцінене в мокроті та рідині з промивання бронхів у астматиків: новий неінвазивний інструмент в епідеміологічних дослідженнях забруднення повітря та астми. Free Radic Biol Med. 1999;27:1448�1454.
38. Найтінгейл JA, Роджерс DF, Barnes PJ. Вплив вдихуваного озону на видихуваний оксид азоту, функцію легенів та індуковану мокротиння у здорових та астматичних суб’єктів. Грудна клітка. 1999;54:1061�1069.
39. Чо А.К., Сіутас С., Мігель А.Х., Кумагай Ю., Шмітц Д.А. та ін. Окисно-відновна активність твердих частинок у повітрі в різних місцях басейну Лос-Анджелеса. Environ Res. 2005;99:40�47.
40. Comhair SA, Thomassen MJ, Erzurum SC. Диференціальна індукція позаклітинної глутатіонпероксидази та синтази оксиду азоту 2 в дихальних шляхах здорових людей, які піддалися впливу 100% O(2) або сигаретного диму. Am J Respir Cell Mol Biol. 2000;23:350�354.
41. Matthay MA, Geiser T, Matalon S, Ischiropoulos H. Оксидантно-опосередковане ураження легень при гострому респіраторному дистрес-синдромі. Crit Care Med. 1999;27:2028�2030.
42. Biaglow JE, Mitchell JB, Held K. Важливість пероксиду та супероксиду в реакції на рентгенівське випромінювання. Int J Radiat Oncol Biol Phys. 1992;22:665�669.
43. Chiu SM, Xue LY, Friedman LR, Oleinick NL. Опосередкована іонами міді сенсибілізація місць прикріплення ядерної матриці до іонізуючого випромінювання. Біохімія. 1993;32:6214�6219.
44. Нараянан П.К., Гудвін Е.Х., Ленерт Б.Є. Альфа-частинки ініціюють біологічне виробництво супероксид-аніонів і перекису водню в клітинах людини. Cancer Res. 1997;57:3963�3971.
45. Tuttle SW, Varnes ME, Mitchell JB, Biaglow JE. Чутливість до хімічних окислювачів і випромінювання в клітинних лініях СНО з дефіцитом активності окисного пентозного циклу. Int J Radiat Oncol Biol Phys. 1992; 22: 671�675.
46. Guo G, Yan-Sanders Y, Lyn-Cook BD, Wang T, Tamae D та ін. Марганець
Експресія генів, опосередкована супероксиддисмутазою, під впливом радіації
адаптивні реакції. Mol Cell Biol. 2003;23:2362�2378.
47. Azzam EI, de Toledo SM, Spitz DR, Little JB. Окислювальний обмін
модулює передачу сигналу та формування мікроядер у спостерігача
клітини з опромінених α-частинок нормальних фібробластів людини. Cancer Res.
2002;62:5436.
48. Ліч JK, Van Tuyle G, Lin PS, Schmidt-Ullrich R, Mikkelsen RB.
Спричинене іонізуючим випромінюванням, залежне від мітохондрій генерування реактивних речовин
кисень/азот. Cancer Res. 2001;61:3894�3901.
49. Dent P, Yacoub A, Fisher PB, Hagan MP, Grant S. MAPK шляхи в
радіаційні реакції. Онкоген. 2003;22:5885�5896.
50. Wei SJ, Botero A, Hirota K, Bradbury CM, Markovina S та ін. Тіоредоксин
ядерна транслокація і взаємодія з окислювально-відновним фактором-1 активує фактор транскрипції AP-1 у відповідь на іонізуюче випромінювання. Cancer Res. 2000;60:6688�6695.
51. Cadet J, Douki T, Gasparutto D, Ravanat JL. Окисне пошкодження ДНК: формування, вимірювання та біохімічні особливості. Mutat Res. 2003;531:5�23.
52. Yokoya A, Cunniffe SM, O'Neill P. Вплив гідратації на індукцію розривів ланцюгів і пошкоджень підстави в плівках плазмідної ДНК шляхом гамма-випромінювання. J Am Chem Soc. 2002;124:8859�8866.
53. Янссен Ю.М., Ван Хаутен Б., Борм П.Д., Моссман Б.Т. Реакція клітин і тканин на окисне пошкодження. Lab Invest. 1993;69:261�274.
54. Iwanaga M, Mori K, Iida T, Urata Y, Matsuo T та ін. Залежна від ядерного фактора каппа B індукція гамма-глутамілцистеїнсинтетази іонізуючою радіацією в клітинах гліобластоми людини T98G. Free Radic Biol Med. 1998;24:1256�1268.
55. Stohs SJ, Bagchi D. Окисні механізми в токсичності іонів металів. Free Radic Biol Med. 1995;18:321�336.
56. Леонард С.С., Харріс Г.К., Ши Х. Окислювальний стрес, індукований металом, і передача сигналу. Free Radic Biol Med. 2004;37:1921�1942.
57. Shi H, Shi X, Liu KJ. Окислювальний механізм токсичності миш'яку та канцерогенезу. Mol Cell Biochem. 2004;255:67�78.
58. Pi J, Horiguchi S, Sun Y, Nikaido M, Shimojo N, Hayashi T. Потенційний механізм порушення утворення оксиду азоту, викликаного тривалим пероральним впливом арсенату у кроликів. Free Radic Biol Med.2003;35:102�113.
59. Рін К., Кавагучі К., Яманака К., Тедзука М., Оку Н., Окада С. Розриви ланцюгів ДНК, викликані диметиларсиновою кислотою, метаболітом неорганічного миш’яку, сильно посилюються супероксидними аніонними радикалами. Biol Pharm Bull. 1995;18:45�58.
60. Waalkes MP, Liu J, Ward JM, Diwan LA. Механізми, що лежать в основі канцерогенезу миш'яку: підвищена чутливість мишей, підданих впливу неорганічного миш'яку під час вагітності. токсикологія. 2004;198:31�38.
61. Шиллер CM, Фаулер BA, Woods JS. Вплив миш'яку на активацію піруватдегідрогенази. Перспектива здоров'я навколишнього середовища. 1977;19:205�207.
62. Монтеріо Х.П., Бечара Е.Х., Абдалла ДСП. Залучення вільних радикалів у неврологічні порфірії та отруєння свинцем. Mol Cell Biochem. 1991;103:73�83.
63. Tripathi RM, Raghunath R, Mahapatra S. Свинець у крові та його вплив на рівні Cd, Cu, Zn, Fe та гемоглобіну у дітей. Sci Total Environ. 2001;277:161�168.
64. Nehru B, Dua R. Вплив харчового селену на нейротоксичність свинцю. J Environ Pathol Toxicol Oncol. 1997;16:47�50.
65. Reid TM, Feig DI, Loeb LA. Мутагенез кисневими радикалами, індукованими металами. Перспектива здоров'я навколишнього середовища. 1994; 102 (додаток 3): 57 61.
66. Кіннула В.Л., Крапо Дж.Д. Супероксиддисмутаза в легенях і легеневих захворюваннях людини. Am J Respir Crit Care Med. 2003;167:1600�1619.
67. Кіннула В.Л. Вироблення та деградація метаболітів кисню під час запальних станів у легенях людини. Препарат Curr спрямований на запальну алергію. 2005;4:465�470.
68. Зелко І.Н., Маріані Т.Д., Фольц Р.Я. Мультигенне сімейство супероксиддисмутази: порівняння структур генів CuZn-SOD (SOD1), Mn-SOD (SOD2) та EC-SOD (SOD3), еволюція та експресія. Free Radic Biol Med. 2002;33:337�349.
69. Кіркман HN, Rolfo M, Ferraris AM, Gaetani GF. Механізми захисту каталази НАДФН. Кінетика і стехіометрія. J Biol Chem. 1999;274:13908�13914.
70. Флох Л. Глутатіонпероксидаза. Основна наука про життя. 1988;49:663�668.
71. Артур мол. Глутатіонпероксидази. Cell Mol Life Sci. 2000;57:1825�1835.
72. Chu FF, Doroshow JH, Esworthy RS. Експресія, характеристика та розподіл у тканинах нової клітинної селензалежної глутатіонпероксидази, GSHPx-GI. J Biol Chem. 1993;268:2571�2576.
73. Comhair SA, Bhathena PR, Farver C, Thunnissen FB, Erzurum SC. Позаклітинна індукція глутатіонпероксидази в легенях з астмою: докази окислювально-відновної регуляції експресії в епітеліальних клітинах дихальних шляхів людини. FASEB J. 2001;15:70�78.
74. Громер С., Уріг С., Бекер К. Система тіоредоксину від науки до клініки. Med Res Rev. 2004; 24:40�89.
75. Kinnula VL, Lehtonen S, Kaarteenaho-Wiik R, Lakari E, P��kk� P та ін. Специфічна клітинна експресія пероксиредоксинів при саркоїдозі легенів і легенів людини. Грудна клітка. 2002;57:157�164.
76. Dubuisson M, Vander Stricht D, Clippe A, Etienne F, Nauser T та ін. Пероксиредоксин 5 людини є пероксинітриредуктазою. FEBS Lett. 2004;571:161�165.
77. Холмгрен А. Антиоксидантна функція систем тіоредоксину та глутаредоксину. Антиоксидно-відновний сигнал. 2000;2:811-820.
78. Дікінсон Д.А., Форман Х.Д. Глутатіон в захисті та сигналізації: уроки малого тіолу. Ann NY Acad Sci. 2002;973:488�504.
79. Сіс Х. Глутатіон та його роль у клітинних функціях. Free Radic Biol Med. 1999;27:916�921.
80. Ladner JE, Parsons JF, Rife CL, Gilliland GL, Armstrong RN. Паралельні еволюційні шляхи глутатіонтрансфераз: структура та механізм каппа-ензиму rGSTK1-1 мітохондріального класу. Біохімія. 2004;43:52�61.
81. Робінсон А., Хаттлі Г.А., Бут Х.С., Борд П.Г. Моделювання та біоінформатичні дослідження глутатіонтрансферази класу каппа людини передбачають нове третє сімейство трансфераз з гомологією до прокаріотичних 2-гідроксихромен-2-карбоксилат-ізомераз. Biochem J. 2004;379:541�552.
82. Jakobsson PJ, Morgenstern R, Mancini J, Ford-Hutchinson A, Persson B. Загальні структурні особливості MAPEGda широко розповсюдженої надсімейства білків, пов'язаних з мембраною, з дуже різними функціями в метаболізмі ейкозаноїдів і глутатіону. Protein Sci. 1999;8:689�692.
83. Hayes JD, Pulford DJ. Сімейство супергенів глутатіон-S-трансферази: регуляція GST та внесок ізоферментів у хіміозахист раку та стійкість до ліків. Crit Rev Biochem Mol Biol. 1995;30:445�600.
84. Армстронг Р.Н. Будова, каталітичний механізм та еволюція глутатіонтрансфераз. Chem Res Toxicol. 1997;10:2�18.
85. Хейс Дж.Д., Маклеллан Л.І. Глутатіон і глутатіонзалежні ферменти представляють собою скоординовано регульований захист від окисного стресу. Free Radic Res. 1999;31:273�300.
86. Sheehan D, Meade G, Foley VM, Dowd CA. Структура, функція та еволюція глутатіонтрансфераз: значення для класифікації членів стародавньої надродини ферментів несавців. Biochem J. 2001; 360: 1�16.
87. Cho SG, Lee YH, Park HS, Ryoo K, Kang KW та ін. Глутатіон S-трансфераза Mu модулює активовані стресом сигнали, пригнічуючи кіназу, що регулює сигнал апоптозу 1. J Biol Chem. 2001;276:12749�12755.
88. Dorion S, Lambert H, Landry J. Активація сигнального шляху p38 тепловим шоком включає дисоціацію глутатіон-S-трансферази Mu від Ask1. J Biol Chem. 2002;277:30792�30797.
89. Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L та ін. Регулювання сигналізації JNK GSTp. EMBO J. 1999;18:1321�1334.
90. Маневич Ю., Файнштейн С.І., Фішер А.Б. Активація антиоксидантного ферменту 1-CYS пероксиредоксину вимагає глутатіонілування, опосередкованого гетеродимеризацією з pGST. Proc Natl Acad Sci US A. 2004; 101:3780�3785.
91. Бункер VW. Вільні радикали, антиоксиданти та старіння. медичні лабораторії 1992;49:299�312.
92. Mezzetti A, Lapenna D, Romano F, Costantini F, Pierdomenico SD та ін. Системний окислювальний стрес і його зв'язок з віком і хворобою. J Am Geriatr Soc. 1996;44:823�827.
93. White E, Shannon JS, Patterson RE. Зв'язок між вітаміном і
вживання добавок кальцію та рак товстої кишки. Біомаркери епідеміолу раку Поперед. 1997; 6: 769-774.
94. Masella R, Di Benedetto R, Vari R, Filesi C, Giovannini C. Нові механізми природних антиоксидантних сполук у біологічних системах: участь глутатіону та пов'язаних з глутатіоном ферментів. J Nutr Biochem. 2005;16:577�586.
95. Curello S, Ceconi C, Bigoli C, Ferrari R, Albertini A, Guarnieri C. Зміни в серцевому статусі глутатіону після ішемії та реперфузії. Experientia. 1985;41:42�43.
96. Ель-Агамі А., Лоу Г.М., МакГарві Діджей, Мортенсен А., Філіп Д.М., Траскотт Т.Г. Хімія радикалів каротиноїдів та антиоксидантні/прооксидантні властивості. Arch Biochem Biophys. 2004;430:37�48.
97. Rice-Evans CA, Sampson J, Bramley PM, Holloway DE. Чому ми очікуємо, що каротиноїди будуть антиоксидантами in vivo? Free Radic Res. 1997;26:381�398.
98. Найлз Р.М. Сигнальні шляхи в хіміопрофілактиці ретиноїдів та лікуванні раку. Mutat Res. 2004;555:81�96.
99. Donato LJ, Noy N. Придушення росту карциноми молочної залози ретиноєвою кислотою: проапоптотичні гени є мішенями для передачі сигналів рецептора ретиноєвої кислоти та клітинного білка II, що зв'язує ретиноєву кислоту. Cancer Res. 2005;65:8193�8199.
100. Ніізума Х, Накамура Ю, Озакі Т, Наканіші Х, Охіра М та ін. Bcl-2 є ключовим регулятором апоптотичної загибелі клітин нейробластоми, спричиненої ретиноєвою кислотою. Онкоген. 2006;25:5046�5055.
101. Dalton TP, Shertzer HG, Puga A. Регуляція експресії генів активним киснем. Ann Rev Pharmacol Toxicol. 1999;39:67�101.
102. Scandalios JG. Геномні реакції на окислювальний стрес. В: Meyers RA, ed. Енциклопедія молекулярної біології клітини та молекулярної медицини. Т. 5. 2-е вид. Вайнхайм, Німеччина: Wiley-VCH; 2004: 489�512.
103. Гош Р., Мітчелл Д.Л. Вплив окисного пошкодження ДНК у промоторних елементах на зв’язування фактора транскрипції. Нуклеїнові кислоти Res. 1999;27:3213�3218.
104. Марієтта С., Гулам Х., Брукс П.Д. Одиночне ураження 8-цикло-50-дезоксиаденозину в коробці ТАТА запобігає зв’язуванню білка, що зв’язує ТАТА, і сильно зменшує транскрипцію in vivo. Ремонт ДНК (Amst). 20;2002:1-967.
105. Джексон А.Л., Чен Р., Леб Л.А. Індукція мікросателітної нестабільності
внаслідок окисного пошкодження ДНК. Proc Natl Acad Sci US A. 1998; 95:12468�12473.
106. Caldecott KW. Взаємодія білка та білка під час відновлення одноланцюгового розриву ДНК ссавців. Biochem Soc Trans. 2003;31:247�251.
107. Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Окислювальне пошкодження ДНК: механізми, мутації та захворювання. FASEB J. 2003;17:1195�1214.
108. Джонс П.Л., Вольф А.П. Взаємозв’язок між організацією хроматину та метилюванням ДНК у визначенні експресії генів. Семіна раку біол. 1999;9:339�347.
109. Джиротті А.В. Механізми перекисного окислення ліпідів. J Free Radic Biol Med. 1985; 1:87�95.
110. Siu GM, Draper HH. Метаболізм малональдегіду in vivo та in vitro. Ліпіди. 1982;17:349�355.
111. Esterbauer H, Koller E, Slee RG, Koster JF. Можлива участь продукту перекисного окислення ліпідів 4-гідроксиноненалю в утворенні флуоресцентних хромоліпідів. Biochem J. 1986; 239: 405-409.
112. Hagihara M, Nishigaki I, Maseki M, Yagi K. Вікові зміни рівнів перекису ліпідів у фракціях ліпопротеїнів сироватки людини. J Gerontol. 1984;39:269�272.
113. Keller JN, Mark RJ, Bruce AJ, Blanc E, Rothstein JD та ін. 4- Гідроксиноненал, альдегідний продукт перекисного окислення мембранних ліпідів, погіршує транспорт глутамату та функцію мітохондрій у синаптосомах. нейронаука. 1997;806:85�96.
114. Uchida K, Shiraishi M, Naito Y, Torii Y, Nakamura Y, Osawa T. Активація сигнальних шляхів стресу кінцевим продуктом перекисного окислення ліпідів. 4-гідрокси-2-ноненал є потенційним індуктором внутрішньоклітинної продукції пероксиду. J Biol Chem. 1999;274:2234�2242.
115. Suc I, Meilhac O, Lajoie-Mazenc I, Vandaele J, Jurgens G, Salvayre R, Negre-Salvayre A. Активація рецептора EGF окисленими ЛПНЩ. FASEB J. 1998;12:665�671.
116. Tsukagoshi H, Kawata T, Shimizu Y, Ishizuka T, Dobashi K, Mori M. 4-Hydroxy-2-nonenal посилює виробництво фібронектину фібробластами легенів людини IMR-90 частково через активацію позаклітинного сигналу, пов'язаного з рецептором епідермального фактора росту. регульований шлях кінази p44/42. Toxicol Appl Pharmacol. 2002;184:127�135.
117. Montuschi P, Collins JV, Ciabattoni G, Lazzeri N, Corradi M, Kharitonov SA, Barnes PJ. Видихуваний 8-ізопростан як біомаркер окисного стресу в легенях in vivo у пацієнтів з ХОЗЛ та здорових курців. Am J Respir Crit Care Med. 2000;162:1175�1177.
118. Morrison D, Rahman I, Lannan S, MacNee W. Епітеліальна проникність, запалення та окислювальний стрес у повітряних просторах курців. Am J Respir Crit Care Med. 1999;159:473�479.
119. Новак Д., Касільський М., Анчак А., П'єтрас Т., Бяласевич П. Підвищений вміст речовин, що реагують на тіобарбітурову кислоту, та перекису водню в конденсаті видихуваного дихання у пацієнтів зі стабільною хронічною обструктивною хворобою легень: відсутність значного впливу куріння сигарет. Респір Мед. 1999;93:389�396.
120. Kelly FJ, Mudway IS. Окислення білка на межі розділу повітря-легені. Амінокислоти. 2003;25:375�396.
121. Dean RT, Roberts CR, Jessup W. Фрагментація позаклітинних і внутрішньоклітинних поліпептидів вільними радикалами. Prog Clin Biol Res. 1985;180:341�350.
122. Кек Р.Г. Використання трет-бутилгідропероксиду як зонда для окислення метіоніну в білках. Анальна біохім. 1996;236:56�62.
123. Девіс К.Дж. Пошкодження та деградація білків кисневими радикалами. I. Загальні аспекти. J Biol Chem. 1987;262:9895�9901.
124. Штадтман Є.Р. Окислення білків, каталізоване іонами металів: біохімічний механізм та біологічні наслідки. Free Radic Biol Med.
1990;9:315.
125. Fucci L, Oliver CN, Coon MJ, Stadtman ER. Інактивація ключових метаболічних ферментів реакціями окислення з змішаною функцією: можливий вплив на обмін білків і старіння. Proc Natl Acad Sci US A. 1983; 80:1521-1525.
126. Stadtman ER, Moskovitz J, Levine RL. Окислення метіонінових залишків білків: біологічні наслідки. Антиоксидний окислювально-відновний сигнал. 2003;5:577�582.
127. Штадтман Є.Р., Левін Р.Л. Опосередковане вільними радикалами окислення вільних амінокислот і амінокислотних залишків у білках. Амінокислоти. 2003;25:207�218.
128. Штадтман Є.Р. Окислення білка при старінні та вікових захворюваннях. Ann NY Acad Sci. 2001;928:22�38.
129. Шактер Е. Кількісна оцінка та значення окислення білка в біологічних зразках. Drug Metab Rev. 2000; 32:307�326.
130. Poli G, Leonarduzzi G, Biasi F, Chiarpotto E. Oxidative stress and cell signaling. Curr Med Chem. 2004;11:1163�1182.
131. Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Фактор росту ендотелію судин (VEGF) та його рецептори. FASEB J. 1999;13:9�22.
132. Sundaresan M, Yu ZX, Ferrans VJ, Sulciner DJ, Gutkind JS та ін. Регуляція утворення активних форм кисню у фібробластах за допомогою Rac1. Biochem J. 1996;318:379-382.
133. Сан Т., Оберлі Л.В. Редокс-регуляція активаторів транскрипції. Free Radic Biol Med. 1996;21:335�348.
134. Klatt P, Molina EP, De Lacoba MG, Padilla CA, Martinez-Galesteo E, Barcena JA, Lamas S. Redox регуляція зв'язування c-Jun ДНК шляхом оборотної S-глутатіоляції. FASEB J. 1999;13:1481�1490.
135. Reynaert NL, Ckless K, Guala AS, Wouters EF, van der Vliet A, Janssen Heininger
Ю.М. Виявлення in situ S-глутатіонілованих білків після дериватізації цистеїну, каталізованої глутаредоксином-1. Biochim Biophys Acta. 2006;1760:380�387.
136. Reynaert NL, Wouters EF, Janssen-Heininger YM. Модуляція глутаредоксину-1
експресія на мишачій моделі алергічного захворювання дихальних шляхів. Am J Respir Cell Mol Biol. 2007;36:147�151.
137. Філомені Г., Ротіліо Г., Чіріоло М.Р. Клітинна передача сигналів і редокс-система глутатіону. Biochem Pharmacol. 2002;64:1057�1064.
138. Pande V, Ramos MJ. Молекулярне розпізнавання 15-дезоксидельта (12,14) простагландину J(2) ядерним фактором-каппа В та іншими клітинними білками. Bioorg Med Chem Lett. 2005;15:4057�4063.
139. Перкінс Н.Д. Інтеграція шляхів клітинної сигналізації з функцією NF-kappaB і IKK. Nat Rev Mol Cell Biol. 2007;8:49�62.
140. Гілмор Т.Д. Вступ до NF-kappaB: гравці, шляхи, перспективи. Онкоген. 2006;25:6680�6684.
141. Hirota K, Murata M, Sachi Y, Nakamura H, Takeuchi J, Mori K, Yodoi J. Різні ролі тіоредоксину в цитоплазмі та в ядрі. Двоступінчастий механізм окислювально-відновної регуляції транскрипційного фактора NF-kappaB. J Biol Chem. 1999;274:27891�27897.
142. Підопічний П.А. Роль комплементу, хемокінів і регуляторних цитокінів при гострому ураженні легенів. Ann NY Acad Sci. 1996;796:104�112.
143. Akira S, Kishimoto A. NF-IL6 і NF-kB в регуляції гена цитокінів. Adv Immunol. 1997;65:1�46.
144. Мейєр М., Шрек Р., Байерле П.А. H2O2 та антиоксиданти мають протилежну дію на активацію NF-каппа B і AP-1 в інтактних клітинах: AP-1 як вторинний фактор, що реагує на антиоксиданти. EMBO J. 1993;12:2005�2015.
145. Abate C, Patel L, Rausher FJ, Curran T. Redox регуляція fos і jun ДНК-зв'язуючої активності in vitro. наук. 1990;249:1157�1161.
146. Galter D, Mihm S, Droge W. Відмінні ефекти дисульфіду глутатіону на ядерні транскрипційні фактори kB та білок-активатор-1. Eur J Biochem. 1994;221:639�648.
147. Hirota K, Matsui M, Iwata S, Nishiyama A, Mori K, Yodoi J. Транскрипційна активність AP-1 регулюється прямим зв'язком між тіоредоксином і Ref-1. Proc Natl Acad Sci US A. 1997; 94: 3633-3638.
Інструмент Find A Practitioner від IFM — це найбільша мережа рефералів у функціональній медицині, створена, щоб допомогти пацієнтам знайти практикуючих функціональних лікарів у будь-якій точці світу. Сертифіковані практикуючі лікарі IFM вказані першими в результатах пошуку, враховуючи їхню широку освіту в галузі функціональної медицини